

Endogenous NO Mediated Stargazin Nitroso Modification in Synaptic Plasticity after Cerebral Ischemia Reperfusion
-
摘要:
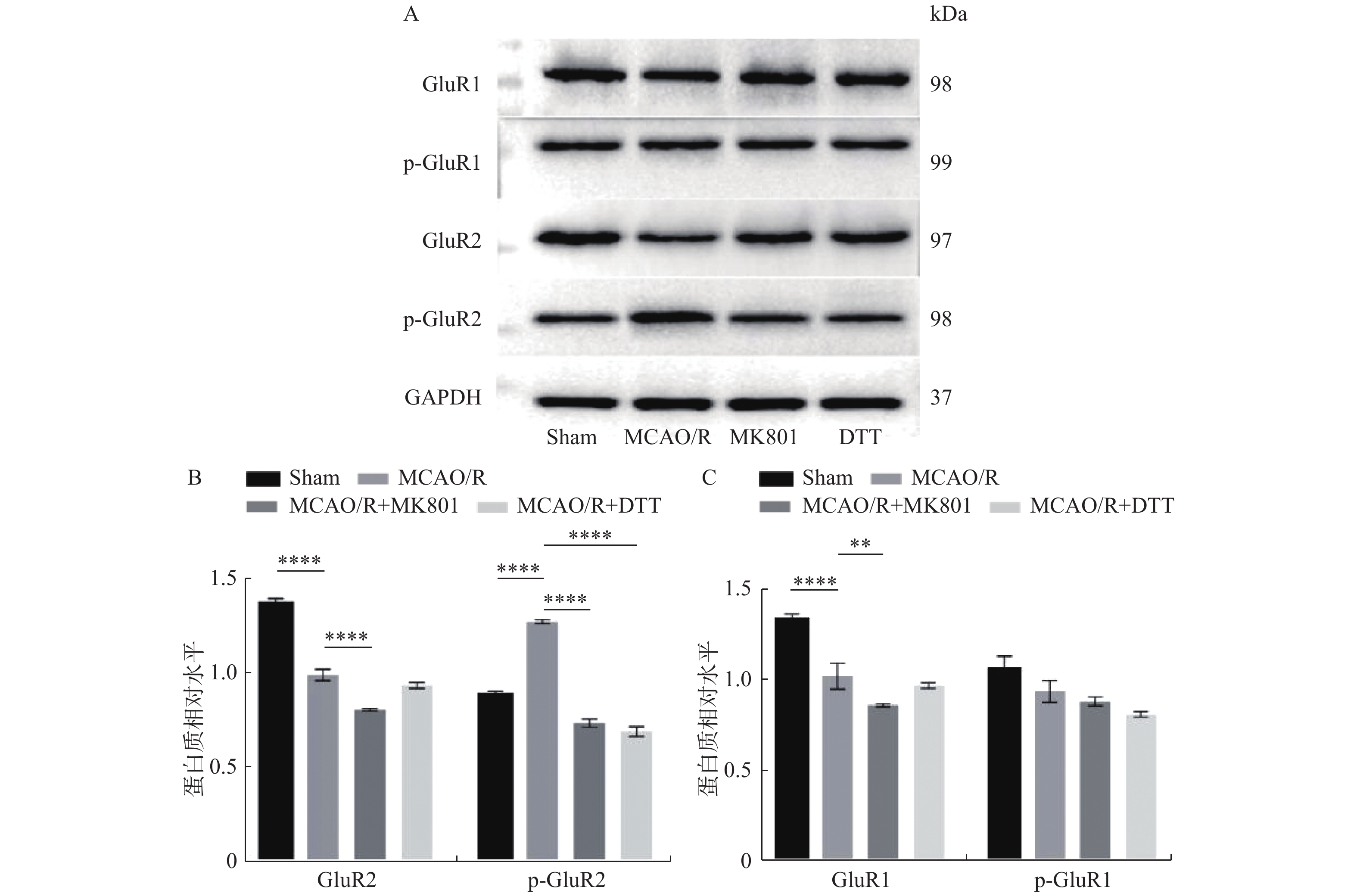
目的 研究Stargazin-亚硝基化修饰在脑缺血再灌注后突触可塑性中的作用,并探讨NO调控AMPAR“Trafficking”的分子机理。 方法 采用线栓法阻塞大脑中动脉复制局灶性脑缺血再灌注(MCAO/R)损伤大鼠作为模型组,分别在每只大鼠MCAO/R模型内给予NMDAR抑制剂MK801、氧化还原剂DTT干预作为实验组。采用mNSS评分标准检测大鼠神经功能,TTC染色检测脑部缺血损伤情况,TUNEL染色检测以及WB检测缺血侧海马神经元凋亡情况,Griess法检测缺血侧海马组织中NO的含量,此外Western Blot检测海马组织中Stargazin-亚硝基化修饰水平以及AMPAR蛋白的表达和活化情况。 结果 给予MK801和DTT处理后MCAO/R模型中Stargazin-亚硝基化修饰水平(P < 0.01)以及NO含量(P < 0.01)下降;AMPAR亚基GluR2磷酸化水平降低(P < 0.0001);抑制Stargazin亚硝基化修饰能够改善MCAO/R引起的神经损伤与凋亡(P < 0.001)。 结论 在MCAO/R模型中,抑制内源性NO和Stargazin亚硝基化水平可促进神经突触重塑,其机制可能是干预了Stargazin辅助蛋白与AMPAR亚基GluR2亲和力有关。 -
关键词:
- 脑缺血再灌注 /
- 一氧化氮(NO) /
- Stargazin-亚硝基化修饰 /
- AMPA 受体“运输” /
- 突触重塑
Abstract:Objective To investigate the role of Stargazin-nitroso modification in synaptic plasticity after cerebral ischemia and reperfusion, and to investigate the molecular mechanism of NO regulating AMPAR “trafficking”. Methods The rat model of focal cerebral ischemia-reperfusion injury was replicated by the middle cerebral artery occlusion (MCAO) method, and the animal model was interfered with NMDAR inhibitor MK801 and oxidative reductant DTT, respectively. mNSS score was used to detect the neurological function of rats. TTC staining and Western Blot were used to detect the ischemic injury of the brain; TUNEL staining was used to detect the apoptosis of hippocampal neurons in the ischemic side; Griess staining was used to detect the content of NO in the ischemic side of hippocampal tissue, and Western Blot was used to detect the stargazin-nitrogenization modification level and AMPAR protein expression and activation in the hippocampal tissue. Results After treatment with MK801 and DTT, the Stargazin-nitroso modification level (P < 0.01) and NO content (P < 0.01) in MCAO/R model were decreased. Phosphorylation of GluR2 in AMPAR subunit was decreased (P < 0.0001); Inhibition of Stargazin’ s nitrosylation modification improved MCAO/ R-induced nerve damage and apoptosis (P < 0.001). Conclusion Inhibition of endogenous NO and Stargazin nitrosylation levels promotes synaptic remodeling in the MCAO/R model, possibly by interfering with the GluR2 affinity between Stargazin helper proteins and AMPAR subunit. -

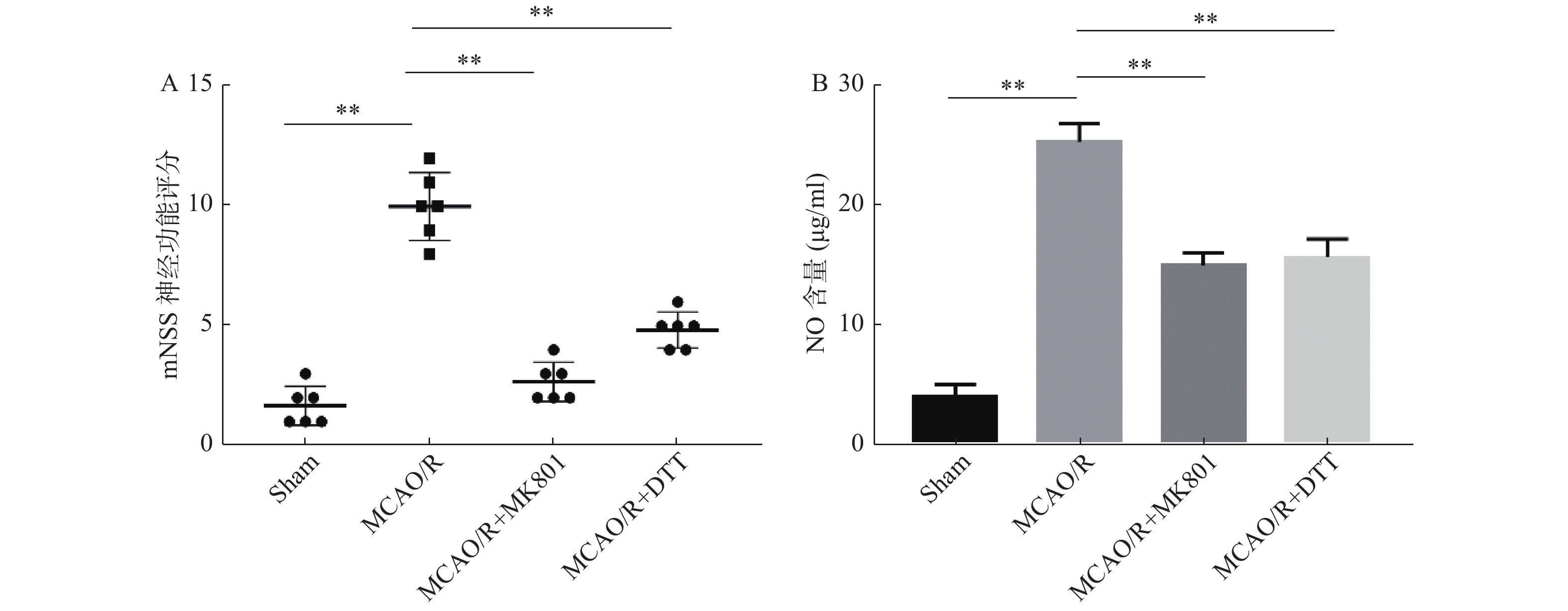
图 1 内源性NO对MCAO/R大鼠神经损伤的影响
A:mNSS神经功能评分;B:以及NO的生成含量。组间两两比较,**P < 0.01。
Figure 1. Effect of endogenous NO on nerve injury in MCAO/R rats

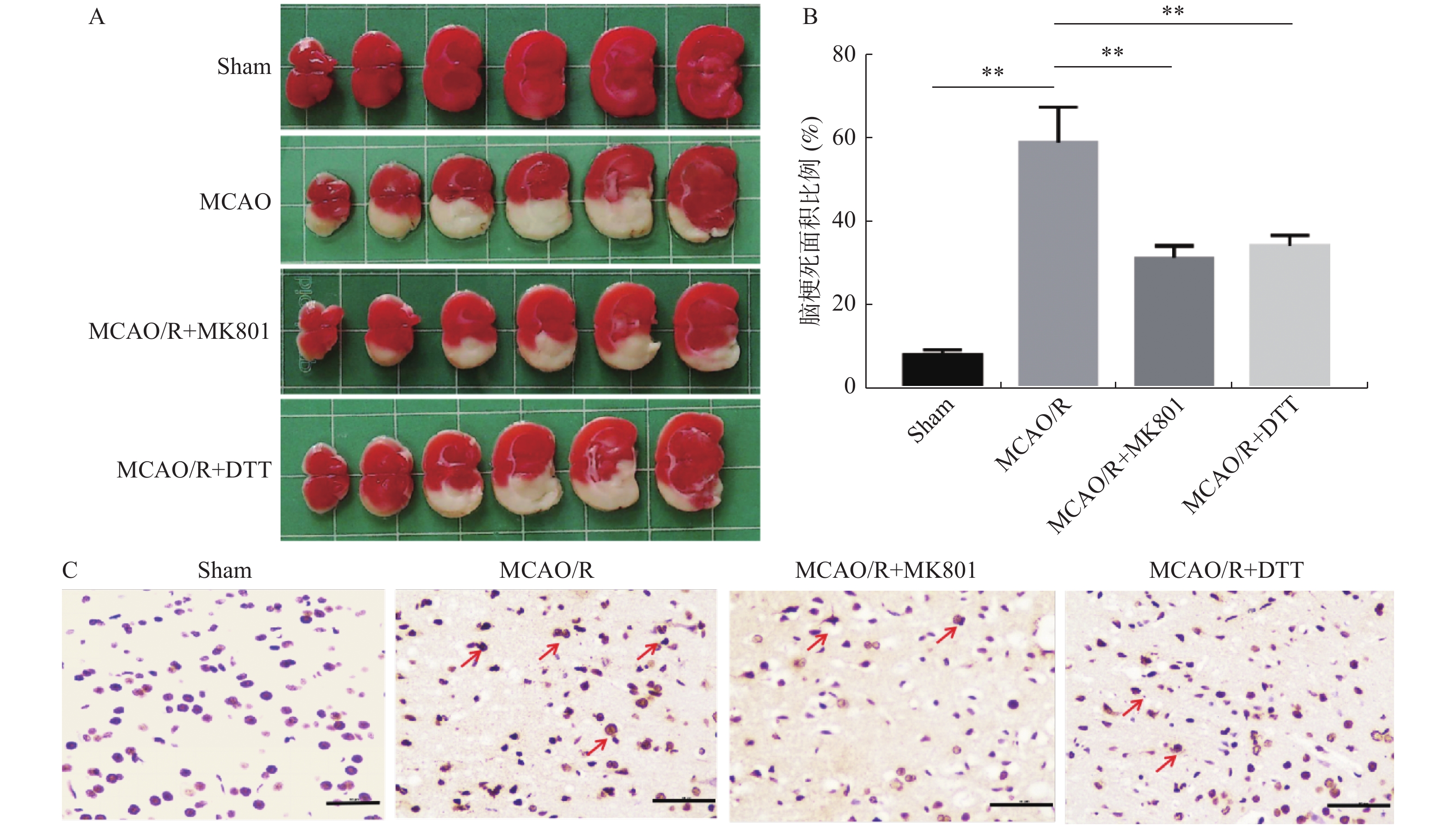
图 2 内源性NO对MCAO/R大鼠脑组织损伤和凋亡的影响
A:脑组织TTC染色结果(白色为梗死区域,横线所示为比例尺50 µm);B:4个组的比较 ;C:脑组织TUNEL染色结果(红色箭头表示凋亡细胞)。 组间两两比较,**P < 0.01。
Figure 2. Effects of endogenous NO on brain tissue injury and apoptosis in MCAO/R rats

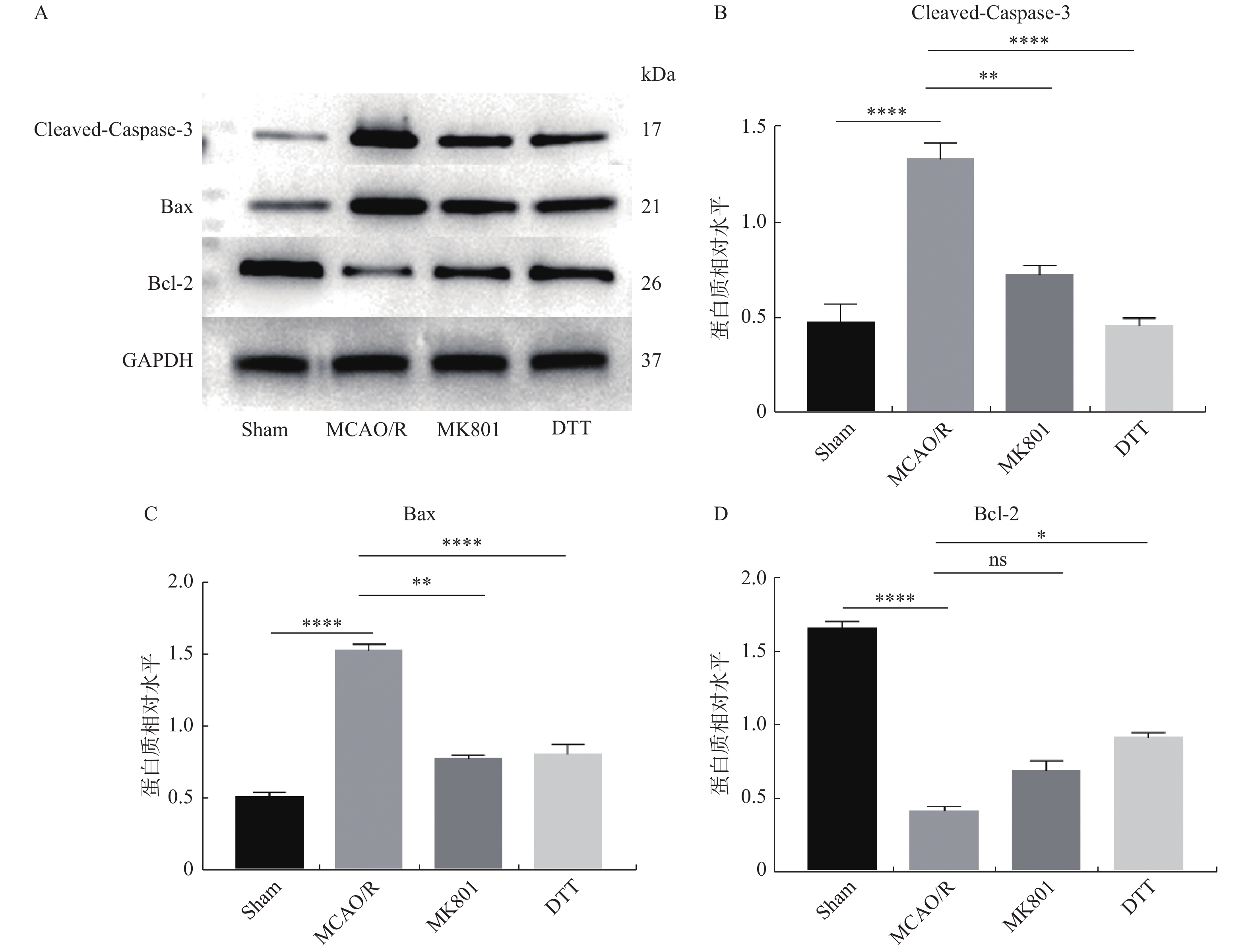
图 3 内源性NO对凋亡相关蛋白的影响
A:Cleaved-Caspase-3、Bax、Bcl-2蛋白的表达;B:Cleaved-Caspase-3蛋白表达水平分析;C:Bax蛋白表达水平分析;D:Bcl-2蛋白表达水平分析。 组间两两比较,*P < 0.05,**P < 0.01,****P < 0.000 1。
Figure 3. Effects of endogenous NO on apoptosis-related proteins

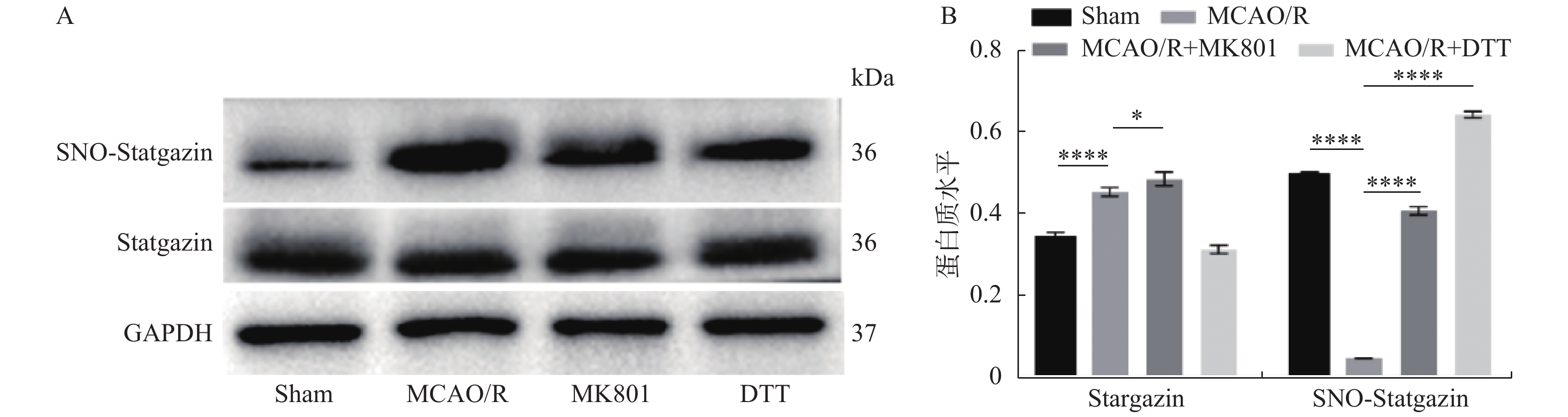
图 4 Western Blot检测海马组织中Stargazin-亚硝基化水平
A:Stargazin总蛋白和亚硝基化水平;B:Stargazin总蛋白和亚硝基化分析。组间两两比较,*P < 0.05,****P < 0.000 1。
Figure 4. Western blot was used to detect the level of Stargazin-nitroso in the hippocampus
-
[1] Nentwhich L M. Diagnosis of Acute Ischemic Stoke[J]. Emergency medicine clinics of North America,2016,34(4):837-859. doi: 10.1016/j.emc.2016.06.008 [2] Nie J,Yang X. Modulation of synaptic plasticity by exercise training as a basis for ischemic stroke rehabilitation[J]. Cellular and molecular neurobiology,2017,37(1):5-16. doi: 10.1007/s10571-016-0348-1 [3] Zhang P,Yu P C,Tsang A H,et al. S-nitrosylation of cyclin-dependent kinase 5 (cdk5) regulates its kinase activity and dendrite growth during neuronal development[J]. The Journal of Neuroscience:The Official Journal of The Society for Neuroscience,2010,30(43):14366-14370. doi: 10.1523/JNEUROSCI.3899-10.2010 [4] Chen J, Hu R, Liao H, et al. A non-ionotropic activity of NMDA receptors contributes to glycine-induced neuroprotection in cerebral ischemia-reperfusion injury[J]. Sci Rep. 2017 7(1): 3575. [5] Martínez Ruiz A,Cadenas S,Lamas S. Nitric oxide signaling:Classical,less classical,and nonclassical mechanisms[J]. Free Radical Biology & Medicine,2011,51(1):17-29. [6] Selvakumar B,Huganir R L,Snyder S H. S-nitrosylation of stargazin regulates surface expression of AMPA-glutamate neurotransmitter receptors[J]. Proceedings of the National Academy of Sciences of the United States of America,2009,106(38):16440-16445. doi: 10.1073/pnas.0908949106 [7] Selvakumar B,Campbell P W,Milovanovic M,et al. AMPA receptor upregulation in the nucleus accumbens shell of cocaine-sensitized rats depends upon S-nitrosylation of stargazin[J]. Neuropharmacology,2014,77(3):28-38. [8] Bell J D,Park E,Ai J,et al. PICK1-mediated GluR2 endocytosis contributes to cellular injury after neuronal trauma[J]. Cell Death and Differentiation,2009,16(12):1665-1680. doi: 10.1038/cdd.2009.106 [9] Diering G H,Huganir R L. The AMPA receptor code of synaptic plasticity[J]. Neuron,2018,100(2):314-329. doi: 10.1016/j.neuron.2018.10.018 [10] 覃启京,覃辉. Stargazin调节AMPA受体功能的相关研究进展[J]. 医学综述,2018,24(18):3564-3569. doi: 10.3969/j.issn.1006-2084.2018.18.006 [11] Tigaret C M,Thalhammer A,Rast G F,et al. Subunit dependencies of N-methyl-D-aspartate (NMDA) receptor-induced alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor internalization[J]. Mol Pharmacol,2006,69(4):1251-1259. [12] Lin D T,Huganir R L. PICK1 and phosphorylation of the glutamate receptor 2 (GluR2) AMPA receptor subunit regulates GluR2 recycling after NMDA receptor-induced internalization[J]. J NEUROSCI,2007,27(50):13903-13908. doi: 10.1523/JNEUROSCI.1750-07.2007 [13] Sun W, Li X, Tang D, et al. Subacute melamine exposure disrupts task-based hippocampal information flow via inhibiting the subunits 2 and 3 of AMPA glutamate receptors expression[J]. . Hum Exp Toxicol. 2021, 40(6): 928-939. [14] Wei Sun,Xiaoliang Li,Dongxin Tang,et al. Subacute melamine exposure disrupts task-based hippocampal information flow via inhibiting the subunits 2 and 3 of AMPA glutamate receptors expression[J]. Human Experimental Toxicology,2021,40(6):928-939. doi: 10.1177/0960327120975821 -
 下载:
下载:
 点击查看大图
点击查看大图
计量
- 文章访问数: 3411
- HTML全文浏览量: 2624
- PDF下载量: 32
- 被引次数: 0
