

Salivary Microbial Profiles of Dental Caries among Lisu Preschool Children in Yunnan Province
-
摘要:
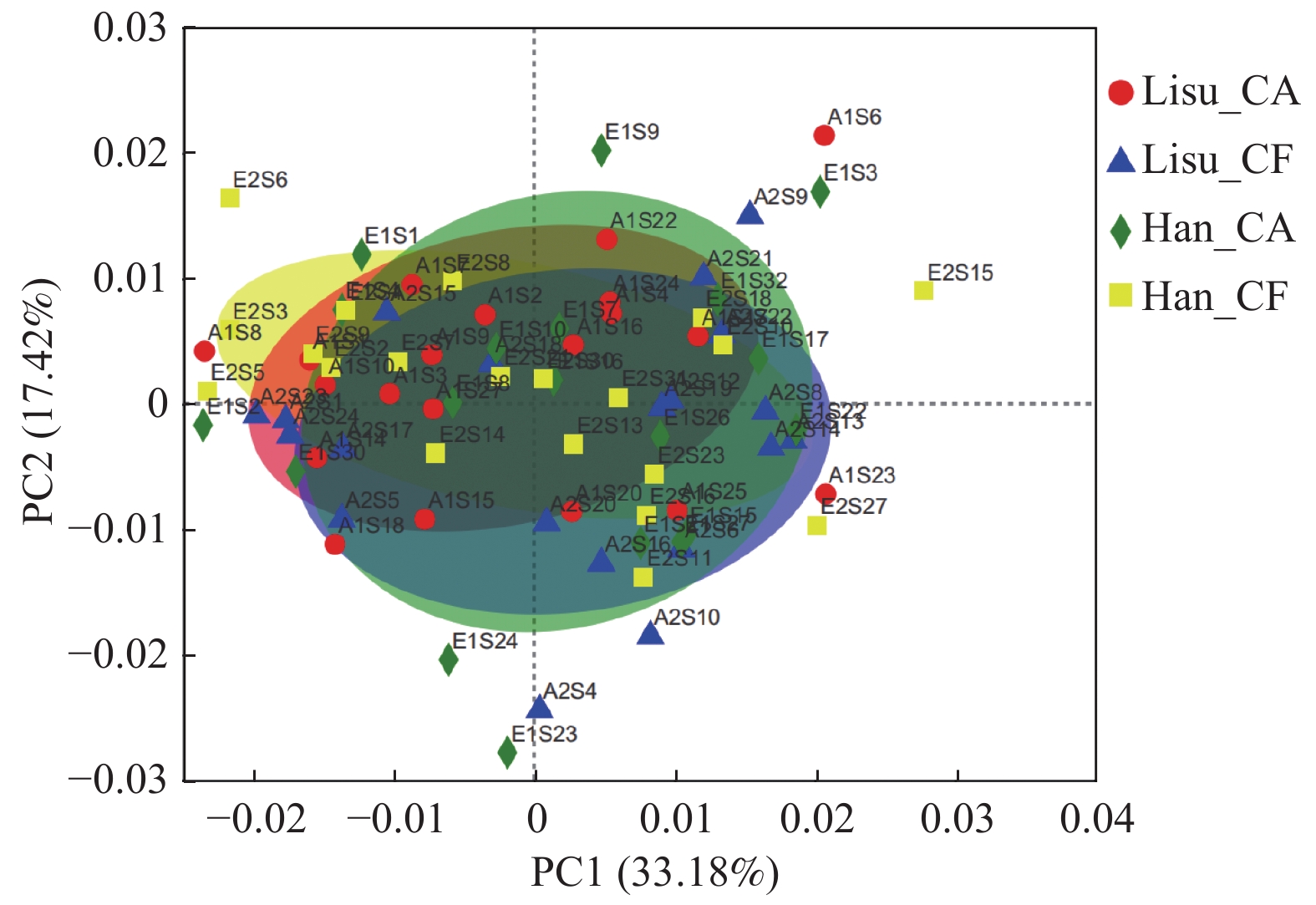
目的 研究云南省5岁傈僳族高龋和无龋儿童的唾液优势菌,并分析与当地汉族儿童的差异。 方法 选取云南省怒江州傈僳族高龋(dmfs≥6)及无龋儿童(dmfs = 0)各20名,同时选取同样的高龋和无龋汉族儿童作为对照,采集唾液样本,运用illumina Hiseq平台对16SrRNA V4区进行单端测序,分析微生物的群落结构和多样性。 结果 基于97%的相似度聚类获得傈僳族、汉族高龋、无龋儿童口腔微生物物种注释(operational taxonomic unit,OTU)数目共3965个,归属于16个门,23个纲,57个目,102个科,202个属;4个组间Alpha多样性及丰富度指数差异无统计学意义(P > 0.05),4组菌群主坐标分析(principal co-ordinates analysis,PCoA分析)差异无统计学意义( P > 0.05);在傈僳族中,高龋组链球菌属( Streptococcus)丰度高于无龋组(P < 0.05),而嗜血杆菌属( Haemophilus)、志贺氏埃希菌属(Escherichia-Shigella)、梭杆菌属(Fusobacterium)丰度低于无龋组(P < 0.05);在高龋组中,傈僳族孪生球菌属( Gemella)丰度高于汉族(P < 0.05),志贺氏埃希菌属( Escherichia-Shigella)丰度低于汉族(P < 0.05)。 结论 云南傈僳族、汉族高龋无龋儿童的唾液微生物群落丰度、多样性、组成相似,但2个民族不同患龋状态有其特异性菌属,造成差异的原因需要进一步研究。 Abstract:Objective To study the salivary predominant bacteria in 5-year-old Lisu children with and without dental caries in Yunnan Province and to analyze the differences with local Han children. Methods 20 Lisu children with high caries (dmfs≥6) and 20 children without caries (dmfs = 0)were selected from Nujiang Prefecture of Yunnan Province. Local Han children were also selected with the same inclusion criteria as comparison. Saliva samples were collected. The 16SrRNA V4 hyper variable region was sequenced using illumina Hisequencing platform. The community structure and diversity of microorganisms were analyzed using Mothur software. Results A total of 3965 oral microbial species annotations (Operational taxonomic unit , OTU) were obtained based on 97% similarity clustering in Lisu and Han Chinese children with and without dental caries, belonging to 16 phyla, 23 classes, 57 orders, 102 families and 202 genera. The differences in Alpha diversity and richness indices among the four groups were not statistically significant (P > 0.05), and the differences in principal co-ordinates analysis (PCoA analysis) among the four groups were not statistically significant ( P > 0.05). The abundance of Streptococcus in the Lisu caries-active group is higher than that of the caries-free group, while the abundance of Haemophilus, Escherichia-Shigella, Fusobacterium, Capnocytophaga, Megasphaera is lower than that of the caries-free group. For caries-active children, the abundance of Gemella was higher in Lisu than in Han, and the abundance of Escherichia-Shigella was lower in Lisu than in Han. Conclusions The diversity, richness and composition of salivary microbial communities were similar among the Lisu and Han children with and without dental caries in Yunnan. However, the dental caries bio-markers of the two ethnic minority were different. The reasons underline the findings need further invesitigation. -
Key words:
- Lisu ethnic minority /
- Children /
- Dental caries /
- 16SrRNA /
- Salivary microorganism
-

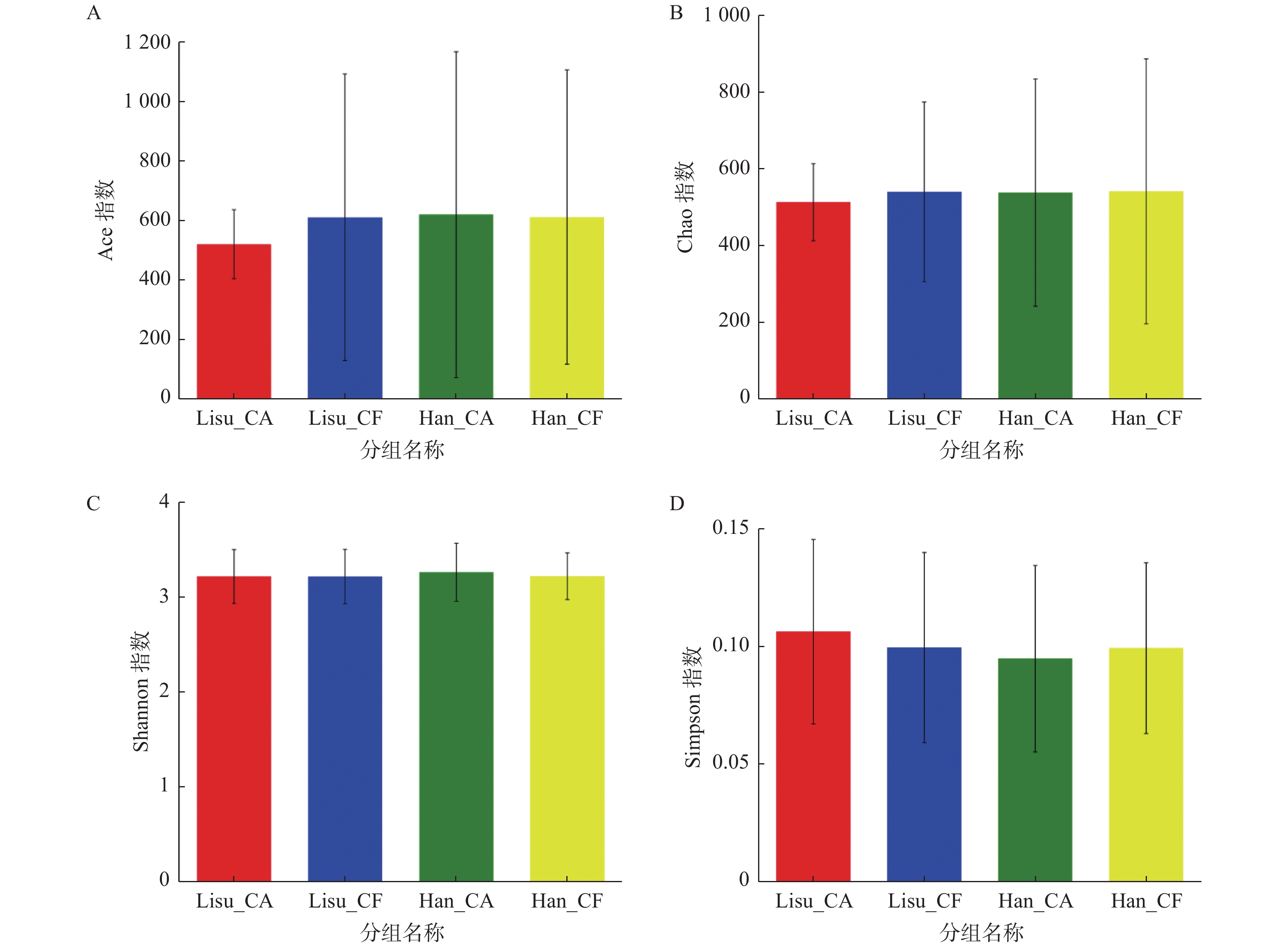
图 2 唾液微生物Alpha多样性指数比较(mean±SD)
A:ACE指数;B:Chao指数;C:Shannon指数;D:Simpson指数
Figure 2. Comparison of salivary microbial Alpha diversity index (mean±SD)
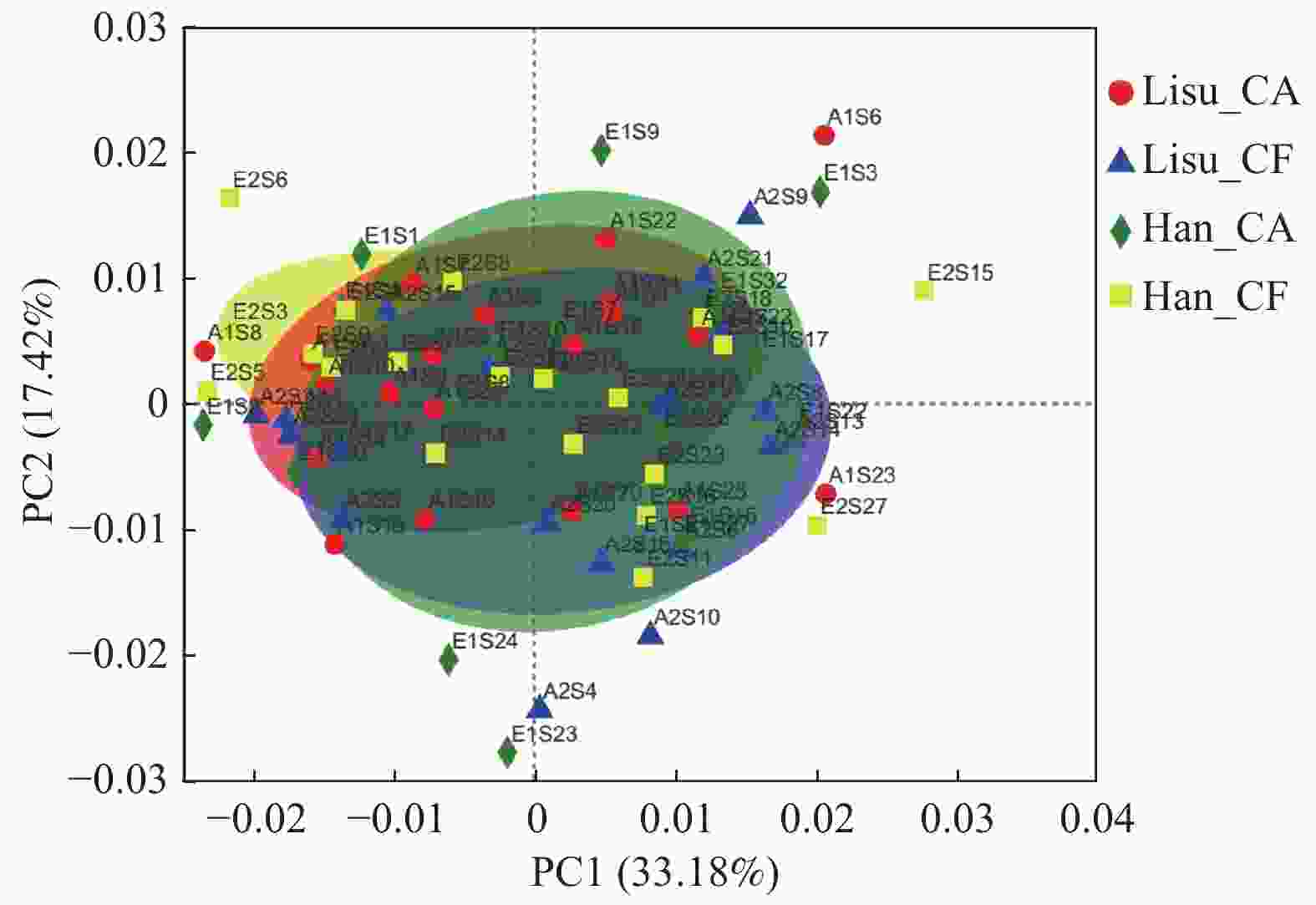
图 3 唾液微生物群落结构的主坐标分析(基于97%相似度的OTU分类水平)
Figure 3. Principal co-ordinates analysis of the salivary microbial community structures (OTU classification level based on 97% similarity)
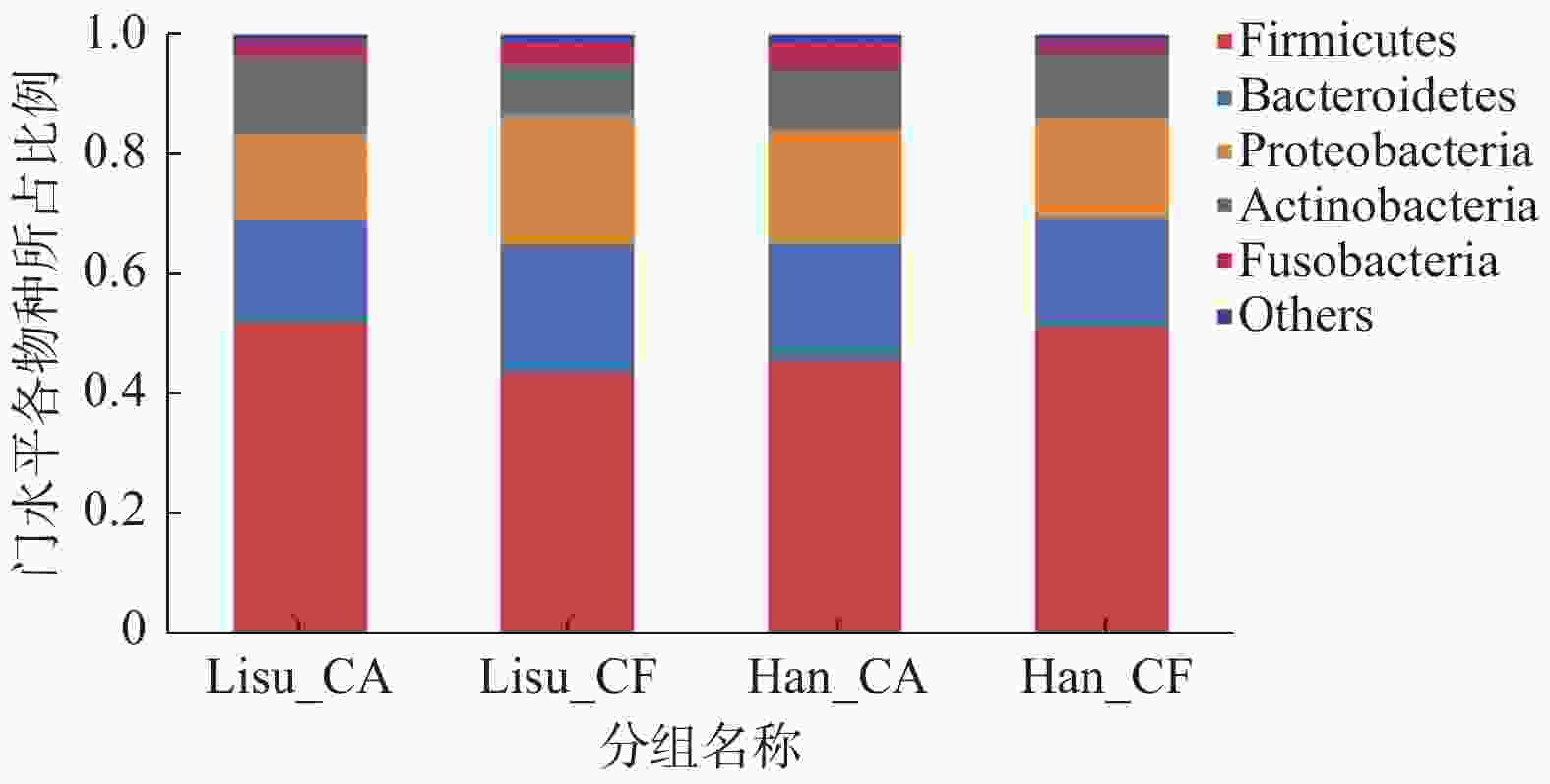
图 4 门水平主要物种分布
Figure 4. The distributions of the predominant bacteria at the phylum level
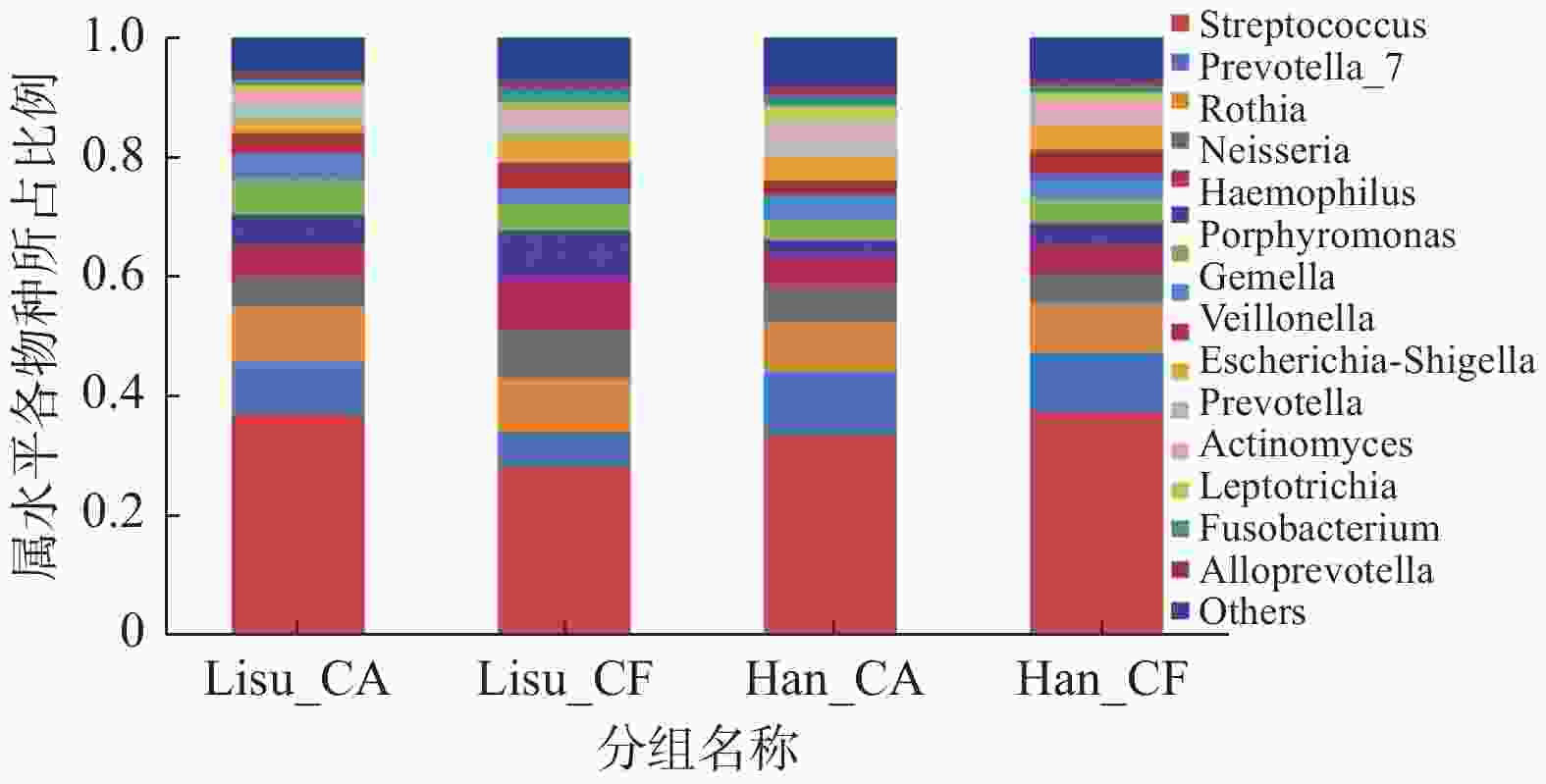
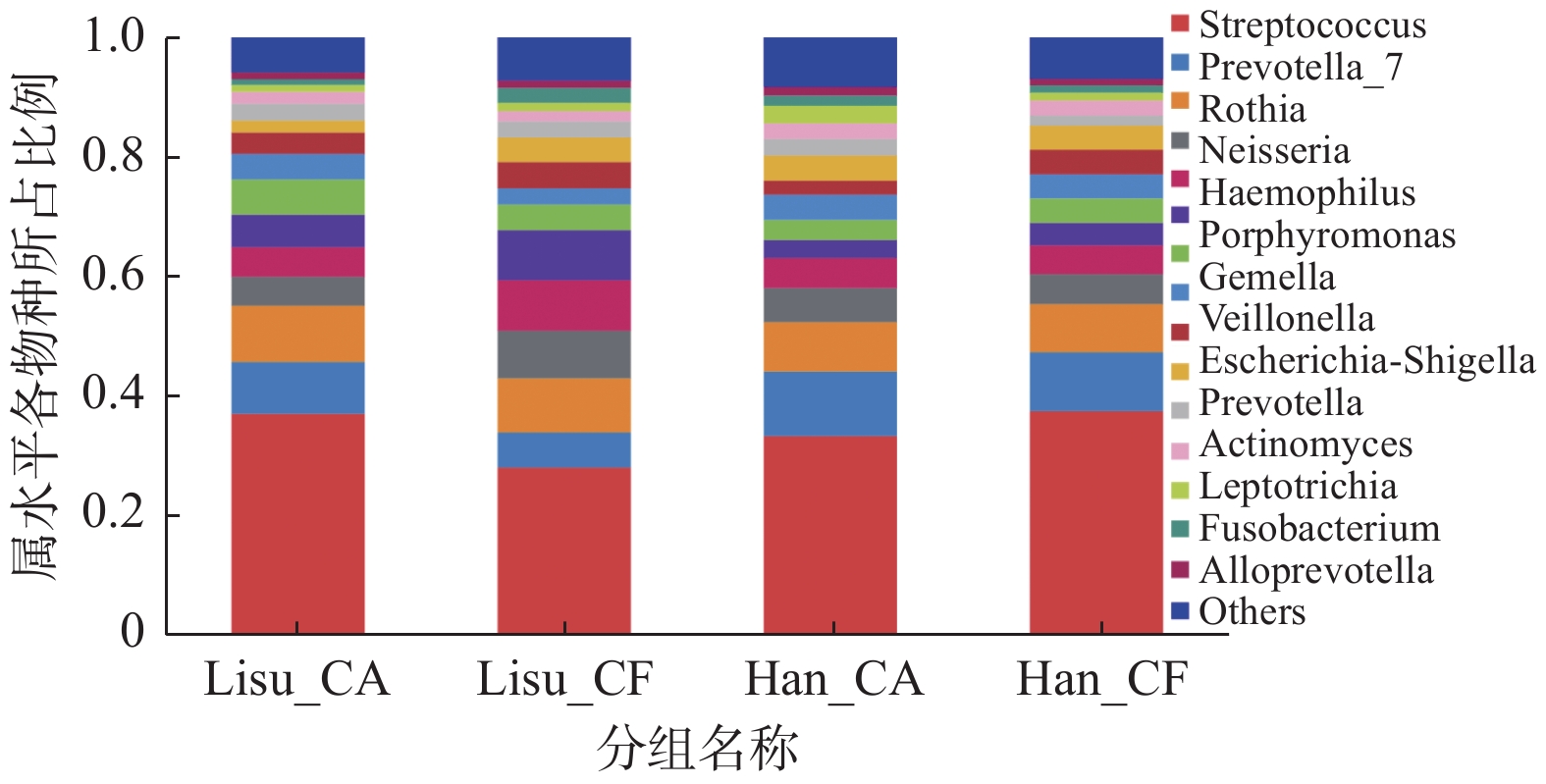
图 5 属水平主要物种分布
Figure 5. The distributions of the predominant bacteria at the genus level

图 6 傈僳族高龋、无龋组唾液微生物群落属分类水平上的组间Wilcoxon秩和检验
Figure 6. Wilcoxon rank-sum test between groups at the genus taxonomic level of salivary microbial communities in the Lisu_CA and Lisu_CF groups

图 7 傈僳族、汉族高龋组唾液微生物群落属分类水平上的组间Wilcoxon秩和检验
Figure 7. Wilcoxon rank-sum test between groups at the genus taxonomic level of salivary microbial communities in the Lisu_CA and Han_CA groups
-
[1] Chenicheri S,R U,Ramachandran R,et al. Insight into oral biofilm:Primary,secondary and residual caries and phyto-challenged solutions[J]. Open Dentistry Journal,2017,11(1):312-333. doi: 10.2174/1874210601711010312 [2] Grigalauskienė R,Slabšinskienė E,Vasiliauskienė I. Biological approach of dental caries management[J]. Stomatologija,2015,17(4):107-112. [3] 王玉霞,周学东,李明云. 韦荣球菌与龋病和链球菌间的关系[J]. 国际口腔医学杂志,2017,44(2):195-199. doi: 10.7518/gjkq.2017.02.016 [4] Alazmah A. Early Childhood Caries: A Review[J]. J Contemp Dent Pract,2017,18(8):732-737. [5] 卢敏,房少华,刘锦桃. 云南16个地区12个民族3~6岁儿童乳牙患龋情况调查分析[J]. 中国妇幼保健,2009,24(1):2. [6] 唐红萍,杨发斌,杨燕槐,等. 云南省15个特有少数民族1~6岁儿童乳牙龋病调查分析[J]. 牙体牙髓牙周病学杂志,2014,24(7):4. [7] Zhang S,Li Y,Liu J,et al. Dental caries status of Lisu preschool children in Yunnan Province,China:a cross-sectional study[J]. BMC Oral Health,2019,19(1):17. doi: 10.1186/s12903-018-0708-y [8] Becker M R,Paster B J,Leys E J,et al. Molecular analysis of bacterial species associated with childhood caries.[J]. Journal of Clinical Microbiology,2002,40(3):1001-1009. doi: 10.1128/JCM.40.3.1001-1009.2002 [9] Zhou X,Li H,Zhu C,et al. Analysis of salivary proteomic biomarkers for the surveillance of changes in high-risk status of early childhood caries[J]. BMC Oral Health,2021,21(1):572. doi: 10.1186/s12903-021-01930-4 [10] Al-Hebshi N N,Baraniya D,Chen T,et al. Metagenome sequencing-based strain-level and functional characterization of supragingival microbiome associated with dental caries in children[J]. J Oral Microbiol,2018,11(1):1557986. [11] Johansson I,Witkowska E,Kaveh B,et al. The microbiome in populations with a low and high prevalence of caries[J]. J Dent Res,2016,95(1):80-86. [12] Magana M,Sereti C,Ioannidis A,et al. Options and limitations in clinical investigation of bacterial biofilms[J]. Clin Microbiol Rev,2018,31(3):e00084-16. [13] Soriano-Lerma A,Pérez-Carrasco V,Sánchez-Marañón M,et al. Influence of 16S rRNA target region on the outcome of microbiome studies in soil and saliva samples[J]. Sci Rep,2020,10(1):13637. doi: 10.1038/s41598-020-70141-8 [14] Leake SL,Pagni M,Falquet L,et al. The salivary microbiome for differentiating individuals: proof of principle[J]. Microbes Infect,2016,18(6):399-405. [15] Kaczor-Urbanowicz K E,Martin Carreras-Presas C,Aro K,et al. Saliva diagnostics - Current views and directions[J]. Exp Biol Med (Maywood),2017,242(5):459-472. doi: 10.1177/1535370216681550 [16] Borkent D,Reardon RJM,McLACHLAN G,et al. A microbiome analysis of equine peripheral dental caries using next generation sequencing[J]. Equine Vet J,2020,52(1):67-75. [17] Choe R,Sim Y F,Hong C H L,et al. Internalizing problems are associated with oral health-related quality of life in early childhood:Outcomes from an Asian multi-ethnic prospective birth cohort[J]. PLoS One,2021,16(8):e0256163. doi: 10.1371/journal.pone.0256163 [18] Pang L,Wang K,Tao Y,et al. A new model for caries risk prediction in teenagers using a machine learning algorithm based on environmental and genetic factors[J]. Front Genet,2021,12:636867. doi: 10.3389/fgene.2021.636867 [19] Du Q,Ren B,He J,et al. Candida albicans promotes tooth decay by inducing oral microbial dysbiosis[J]. ISME J,2021,15(3):894-908. doi: 10.1038/s41396-020-00823-8 [20] Jiang S,Gao X,Jin L,Lo EC. Salivary microbiome diversity in caries-free and caries-affected children[J]. Int J Mol Sci,2016,17(12):1978. doi: 10.3390/ijms17121978 [21] Xu H,Tian J,Hao W,et al. Oral microbiome shifts from caries-free to caries-affected status in 3-year-old Chinese children:A longitudinal study[J]. Front Microbiol,2018,9:2009. doi: 10.3389/fmicb.2018.02009 [22] Xiao C,Ran S,Huang Z,et al. Bacterial diversity and community structure of supragingival plaques in adults with dental health or caries revealed by 16S pyrosequencing[J]. Front Microbiol,2016,7:1145. [23] Zhang Y,Huang S,Jia S,et al. The predictive power of saliva electrolytes exceeds that of saliva microbiomes in diagnosing early childhood caries[J]. J Oral Microbiol,2021,13(1):1921486. doi: 10.1080/20002297.2021.1921486 [24] Conrads G,About I. Pathophysiology of dental caries[J]. Monogr Oral Sci,2018,27:1-10. [25] Boisen G,Davies J R,Neilands J. Acid tolerance in early colonizers of oral biofilms[J]. BMC Microbiol,2021,21(1):45. doi: 10.1186/s12866-021-02089-2 [26] Chen W,Jiang Q,Yan G,Yang D. The oral microbiome and salivary proteins influence caries in children aged 6 to 8 years[J]. BMC Oral Health,2020,20(1):295. doi: 10.1186/s12903-020-01262-9 [27] Li Y,Zou C G,Fu Y,et al. Oral microbial community typing of caries and pigment in primary dentition[J]. Bmc Genomics,2016,17(1):558. doi: 10.1186/s12864-016-2891-z [28] Zhang Y,Zhu C,Feng X,Chen X. Microbiome variations in preschool children with halitosis[J]. Oral Dis,2021,27(4):1059-1068. doi: 10.1111/odi.13603 [29] Belotserkovsky I,Sansonetti P J. Shigella and Enteroinvasive Escherichia Coli[J]. Curr Top Microbiol Immunol,2018,416:1-26. -
 下载:
下载:



 点击查看大图
点击查看大图
计量
- 文章访问数: 4137
- HTML全文浏览量: 2221
- PDF下载量: 29
- 被引次数: 0
