

Clinical Characteristics and Genetic Testing of A Pedigree with Early-onset Parkinson's Disease Caused by PRKN Gene Mutation
-
摘要:
目的 探讨2个常染色体隐性遗传早发型帕金森病(autosomal recessive early-onset parkinsonism,AREP)家系中2名患者的临床特征及基因突变情况。 方法 对2个中国汉族家庭中共2名患者进行临床资料的收集和基因突变分析。使用靶区捕获和高通量测序筛选与帕金森病(Parkinson’s disease,PD)、震颤、脊髓小脑性共济失调和肌张力障碍等疾病相关的基因;应用多重连接依赖探针扩增(multiples ligation-dependent probe amplification,MLPA)法检测SNCA、LRRK2、PARK2、PINK1、PARK7、ATP13A2、UCHL1、GCH1等基因外显子的重排和大缺失突变。 结果 2名临床确诊为PD的患者表现出明显的临床及遗传异质性。基因检测发现家系1的患者存在PRKN基因2号外显子杂合缺失变异和c.619G > T/p.Glu207Ter*杂合变异2种突变,该复合杂合变异与疾病存在家系共分离。家系2的患者存在PRKN基因3~4号外显子纯合缺失变异,且存在LRRK2基因c.4827+6T > A杂合变异及PINK1基因c.1474C > T/p.Arg492* 杂合变异;生物信息学分析发现LRRK2基因的c.4827+6T > A变异可能导致其剪切改变。 结论 PRKN基因突变所致的早发型帕金森病临床表现及基因突变形式多样;AREP患者可能同时存在多个PD基因致病突变,且其临床发病年龄更早,症状更重更复杂,病情进展更快。 Abstract:Objective To investigate the clinical characteristics and gene mutations of 2 patients in 2 families of autosomal recessive early-onset Parkinson’s disease (AREP). Methods Clinical data and gene mutation analysis were performed on 2 patients from 2 Chinese Han families. Target capture and high-throughput sequencing were used to screen genes related to Parkinson’s disease (PD), tremor, spinocerebellar ataxia, and dystonia; Multiple ligation-dependent probe amplification (MLPA) was used to detect the rearrangement and large deletion mutations of SNCA, LRRK2, PARK2, PINK1, PARK7, ATP13A2, UCHL1, GCH1 gene exons. Results 2 patients with clinically confirmed PD showed the obvious clinical and genetic heterogeneity. Gene detection found that there were two mutations in the PRKN gene exon 2 heterozygous deletion mutation and c.619G > T/p.Glu207Ter * heterozygous mutation in the patient of family 1. The compound heterozygous mutation was pedigree cosegregated in the family. The patients of pedigree 2 had homozygous deletion mutation in exon 3-4 of PRKN gene, and had heterozygous mutation in LRRK2 gene c.4827+6T > A, and heterozygous mutation in PINK1 gene c.1474C > T/p.Arg492*; Bioinformatics analysis found that the c.4827+6T > A mutation of LRRK2 gene may lead to its shear change. Conclusion The clinical manifestations and gene mutations of early-onset Parkinson’s disease caused by PRKN gene mutations are diverse; AREP patients may have multiple PD gene pathogenic mutations at the same time, and their clinical onset age is earlier, the symptoms are more severe and complex, and the disease progresses are faster. -
Key words:
- Early onset Parkinson’s disease /
- PRKN gene /
- LRRK2 gene /
- PINK1 gene
-
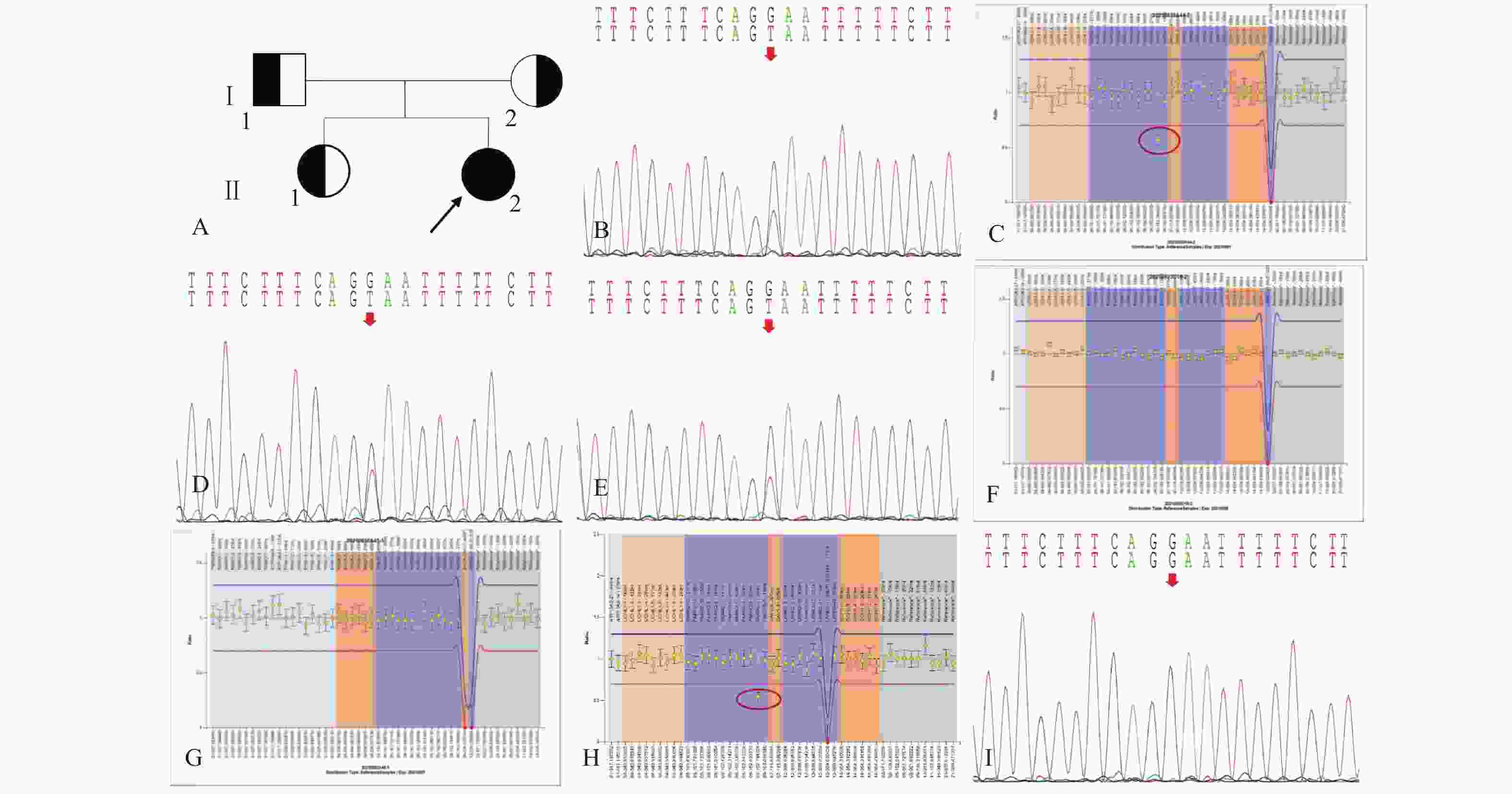
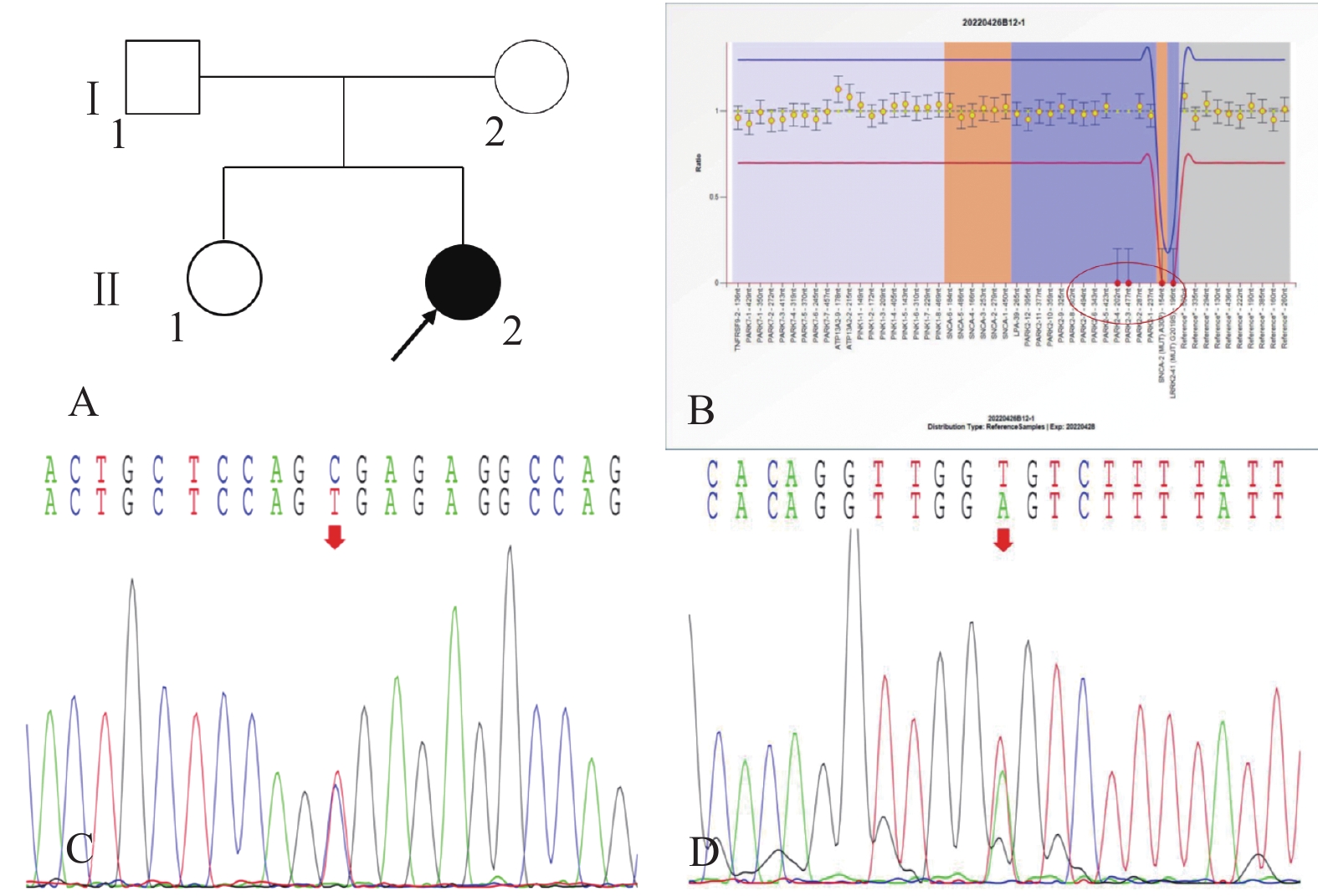
图 1 家系1的系谱图及基因测序图
A:家系1系谱图;B:患者(先证者,II-2)存在PRKN基因c.619G>T/p.Glu207Ter*杂合变异;C:患者(先证者,II-2)存在PRKN基因2号外显子杂合缺失;D:患者父亲(I-1)携带PRKN基因c.619G>T/p.Glu207Ter*杂合变异;E:患者姐姐(II-1)携带c.619G>T/p.Glu207Ter*杂合变异;F:患者父亲(I-1)不携带PRKN基因2号外显子杂合缺失;G:患者姐姐(II-1)不携带PRKN基因2号外显子杂合缺失;H:患者的母亲(II-2)携带PRKN基因2号外显子杂合缺失;I:患者的母亲(II-2)不携带PRKN基因c.619G>T/p.Glu207Ter*变异。
Figure 1. Pedigree and gene sequencing of family 1
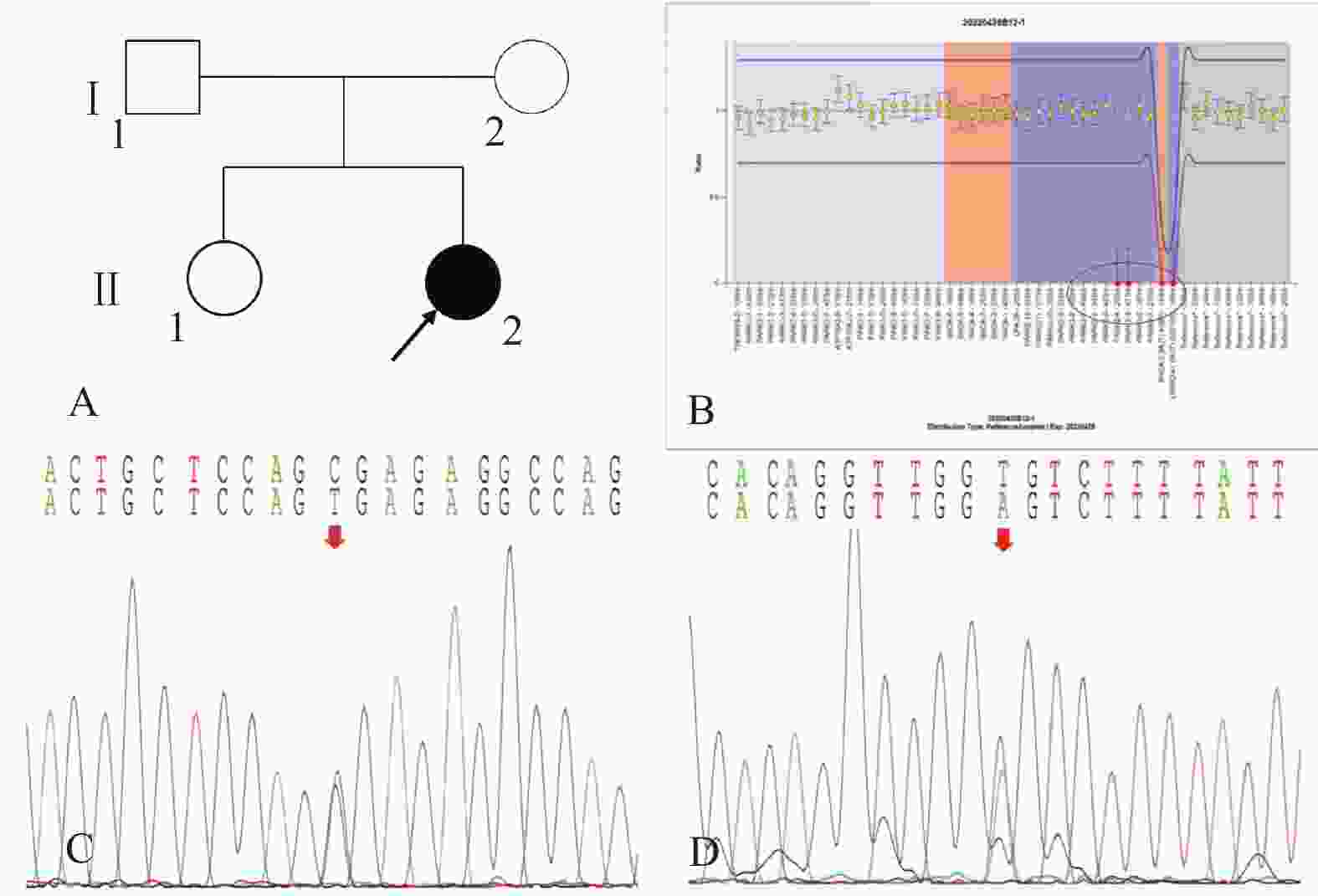
图 2 家系2的系谱图及基因测序图
A:家系2系谱图;B:患者(先证者,II-2)存在PRKN基因3-4号外显子纯合缺失变异; C:患者(先证者,II-2)存在PINK1基因c.1474C>T/p.Arg492* 杂合无义变异;D:患者(先证者,II-2)存在 LRRK2基因c.4827+6T>A杂合剪接型变异。
Figure 2. Pedigree and gene sequencing of family 2
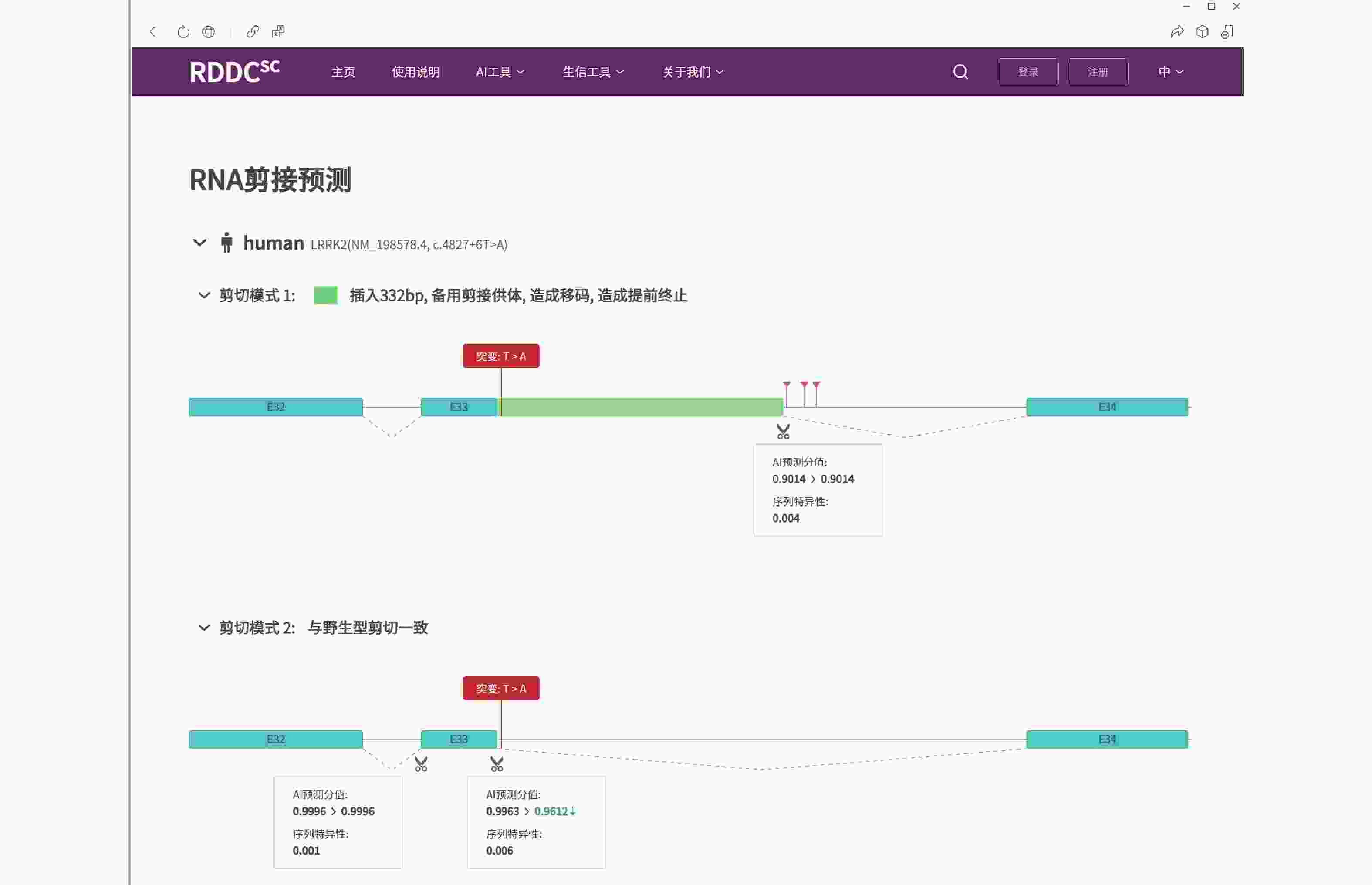
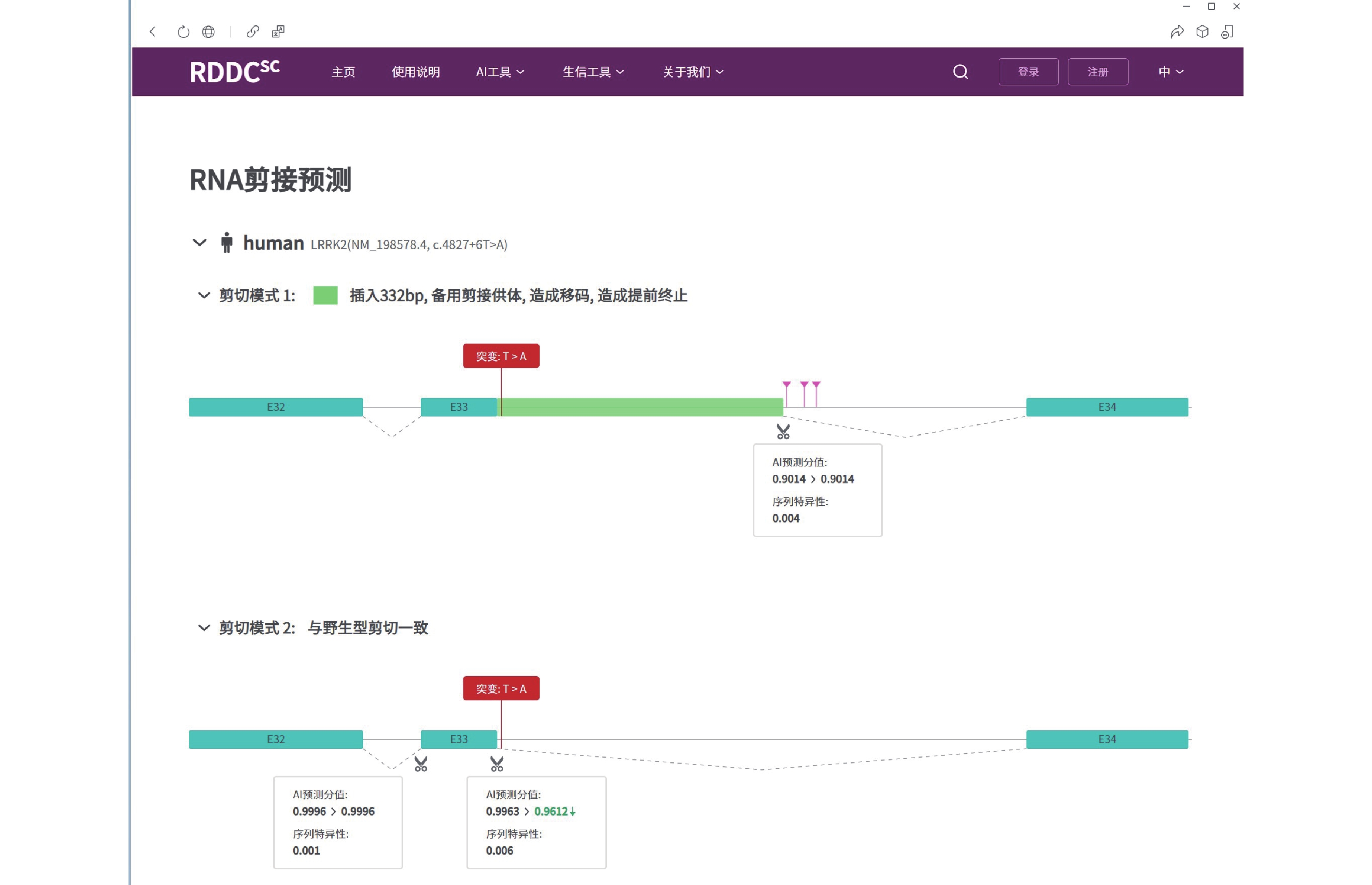
图 3 LRRK2基因c.4827+6T>A变异的剪接预测(https://rddc.tsinghua-gd.org/tools/patho-predic/result?uuid=005c2fe7-8ebf-4489-a3b0-802b42aeabfb)
LRRK2基因的c.4827+6T>A变异可能导致剪切改变,插入332 bp碱基造成移码而提前终止翻译成截短蛋白。
Figure 3. Splicing prediction of LRRK2 gene c.4827+6T>A mutation
表 1 2名EOPD患者的临床特征及基因突变结果
Table 1. Clinical characteristics and gene mutation results of 2 EOPD patients
临床特征 家系1患者 家系2患者 性别 女 女 发病年龄(岁) 29 28 疾病持续时间(年) 3 13 静息震颤 + ++ 肌强直 + ++ 运动缓慢 + ++ 姿势不稳 − ++ 剂末现象 − + 开期/关期 − + 自主神经功能障碍 − ++ 快速动眼睡眠行为障碍 − + 嗅觉障碍 − + 起病时肌张力障碍 − + 反射亢进 − + 精神症状 − + 对左旋多巴反应性 + + Hoehn-Yahr 1.5 2.5 UPDRS III score 11 29 MMSE 25 20 HAMD 4 20 HAMA 2 8 PDQ39 4 18 基因检测结果 PRKN基因的复合杂合突变:
2号外显子杂合缺失变异、
c.619G>T/p.Glu207Ter*杂合变异PRKN基因:3-4号外显子纯合缺失变异;
LRRK2基因:c.4827+6T > A杂合变异;
PINK1基因:c.1474C>T/p.Arg492* 杂合变异“+”表示“存在”,“−”表示“不存在”;UPDRS III:统一帕金森病分级量表第三部分;NMSS:帕金森病非运动症状评分;MMSE:简易精神状态检查;HAMD:汉密尔顿抑郁量表;HAMA:汉密尔顿焦虑量表;PDQ39:帕金森病问卷。  下载: 导出CSV
下载: 导出CSV
-
[1] Tysnes O B,Storstein A. Epidemiology of parkinson's disease[J]. J Neural Transm (Vienna),2017,124(8):901-905. doi: 10.1007/s00702-017-1686-y [2] Simon D K,Tanner C M,Brundin P. Parkinson disease epidemiology,pathology,genetics,and pathophysiology[J]. Clin Geriatr Med,2020,36(1):1-12. doi: 10.1016/j.cger.2019.08.002 [3] Hui Ye,Laurie A Robak,Meigen Yu,et al. Genetics and Pathogenesis of Parkinson’s Syndrome[J]. Annu Rev Pathol,2023,18(1):95-121. [4] Max Borsche,Sandro L Pereira,Christine Klein,et al. Mitochondria and parkinson’s disease:Clinical,molecular,and translational aspects[J]. J Parkinsons Dis,2021,11(1):45-60. doi: 10.3233/JPD-201981 [5] Blauwendraat C,Nalls M A,Singleton A B. The genetic architecture of parkinson's disease[J]. Lancet Neurol,2020,19(2):170-178. doi: 10.1016/S1474-4422(19)30287-X [6] Ascherio A,Schwarzschild M A. The epidemiology of Parkinson's disease: Risk factors and prevention[J]. Lancet Neurol,2016,15(12):1257-1272. doi: 10.1016/S1474-4422(16)30230-7 [7] Tolosa E,Vila M,Klein C,et al. LRRK2 in Parkinson disease: Challenges of clinical trials[J]. Nat Rev Neurol,2020,16(2):97-107. doi: 10.1038/s41582-019-0301-2 [8] Kasten M,Hartmann C,Hampf J,et al. Genotype-phenotype relations for the Parkinson’s disease genes Parkin,PINK1,DJ1: MDSGene systematic review[J]. Mov Disord,2018,33(5):730-741. doi: 10.1002/mds.27352 [9] Periquet M, Latouche M, Lohmann E, et al. Parkin mutations are frequent in patients with isolated early-onset parkinsonism[J]. Brain, 2003, 126(Pt 6): 1271-1278. [10] Zhao Yuwen,Qin Lixia,Pan Hongxu,et al. The role of genetics in Parkinson’s disease:A large cohort study in Chinese mainland population[J]. Brain,2020,143(7):2220-2234. doi: 10.1093/brain/awaa167 [11] Shimura H,Hattori N,Si K,et al. Familial parkinson disease gene product,parkin,is a ubiquitin-protein ligase[J]. Nat Genet,2000,25(3):302-305. doi: 10.1038/77060 [12] Guo J F,Zhang X W,Nie L L,et al. Mutation analysis of parkin,PINK1 and DJ-1 genes in Chinese patients with sporadic early onset parkinsonism[J]. J Neurol,2010,257(7):1170-1175. doi: 10.1007/s00415-010-5485-8 [13] Dunhui Li,May T Aung-Htut,Kristin A Ham,et al. A splice intervention therapy for autosomal recessive juvenile Parkinson’s disease arising from Parkin mutations[J]. Int J Mol Sci,2020,21(19):7282-7296. doi: 10.3390/ijms21197282 [14] 中华医学会神经病学分会帕金森病及运动障碍学组,中国医师协会神经内科医师分会帕金森病及运动障碍学组. 中国帕金森病的诊断标准(2016版)[J]. 中华神经科杂志,2016,49(4):268-271. doi: 10.3760/cma.j.issn.1006-7876.2016.04.002 [15] 中华医学会神经病学分会帕金森病及运动障碍学组,中国医师协会神经内科医师分会帕金森病及运动障碍学组. 早发型帕金森病的诊断与治疗中国专家共识[J]. 中华神经医学杂志,2021,20(2):109-116. [16] Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism[J]. Nature, 1998, 392(6676): 605-608. [17] Ge P,Dawson V L,Dawson T M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease[J]. Mol Neurodegener,2020,15(20):1-18. [18] Li Jie,Lai Mengyu,Zhang Xixi,et al. PINK1-parkin-mediated neuronal mitophagy deficiency in prion disease[J]. Cell Death Dis,2022,13(2):162-173. doi: 10.1038/s41419-022-04613-2 -
 点击查看大图
点击查看大图

计量
- 文章访问数: 4087
- HTML全文浏览量: 2958
- PDF下载量: 36
- 被引次数: 0
