

Bioinformatics-Based Analysis of the Roles of MX1,IFI44, and STAT1 in Lupus Nephritis
-
摘要:
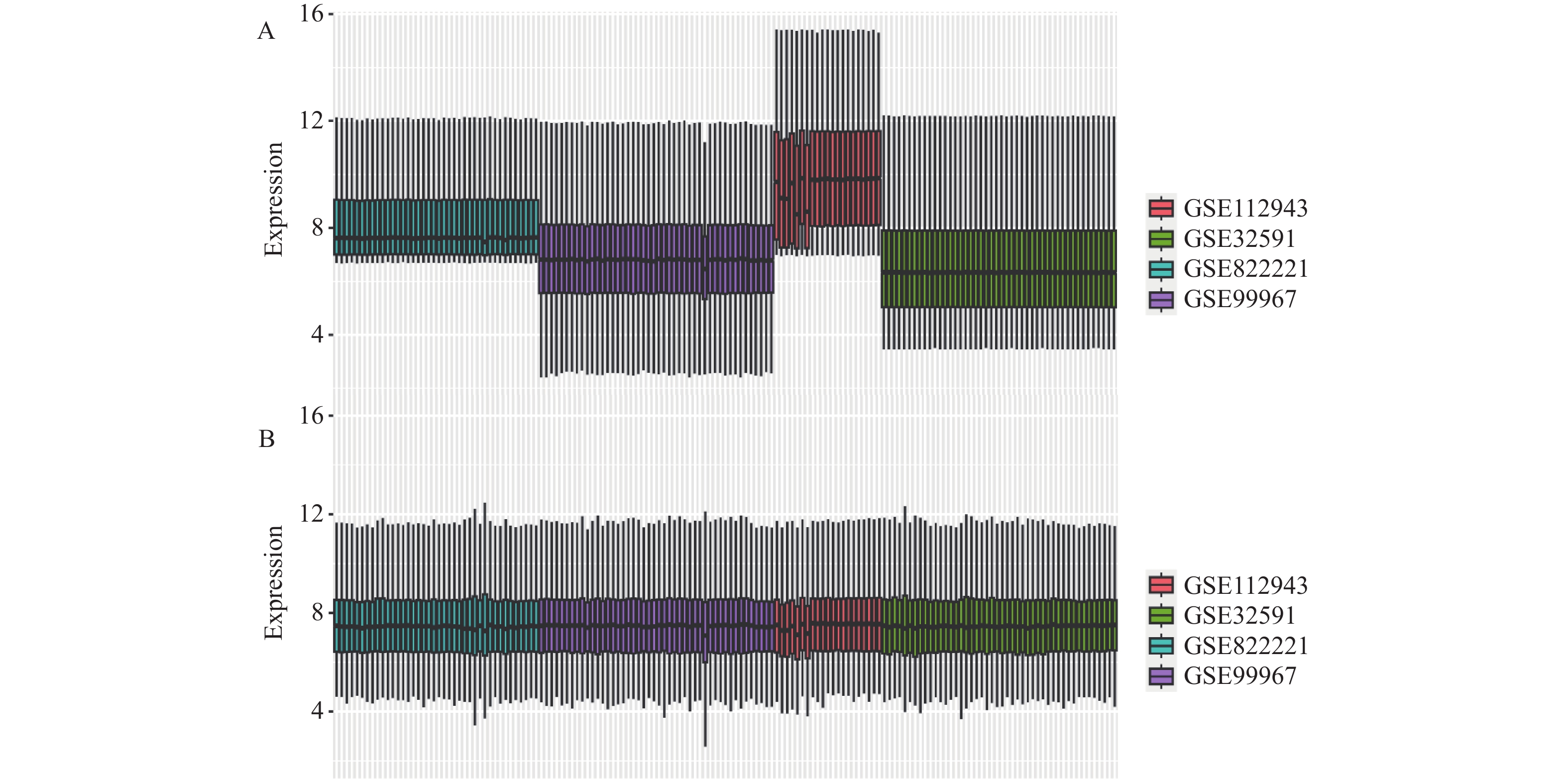
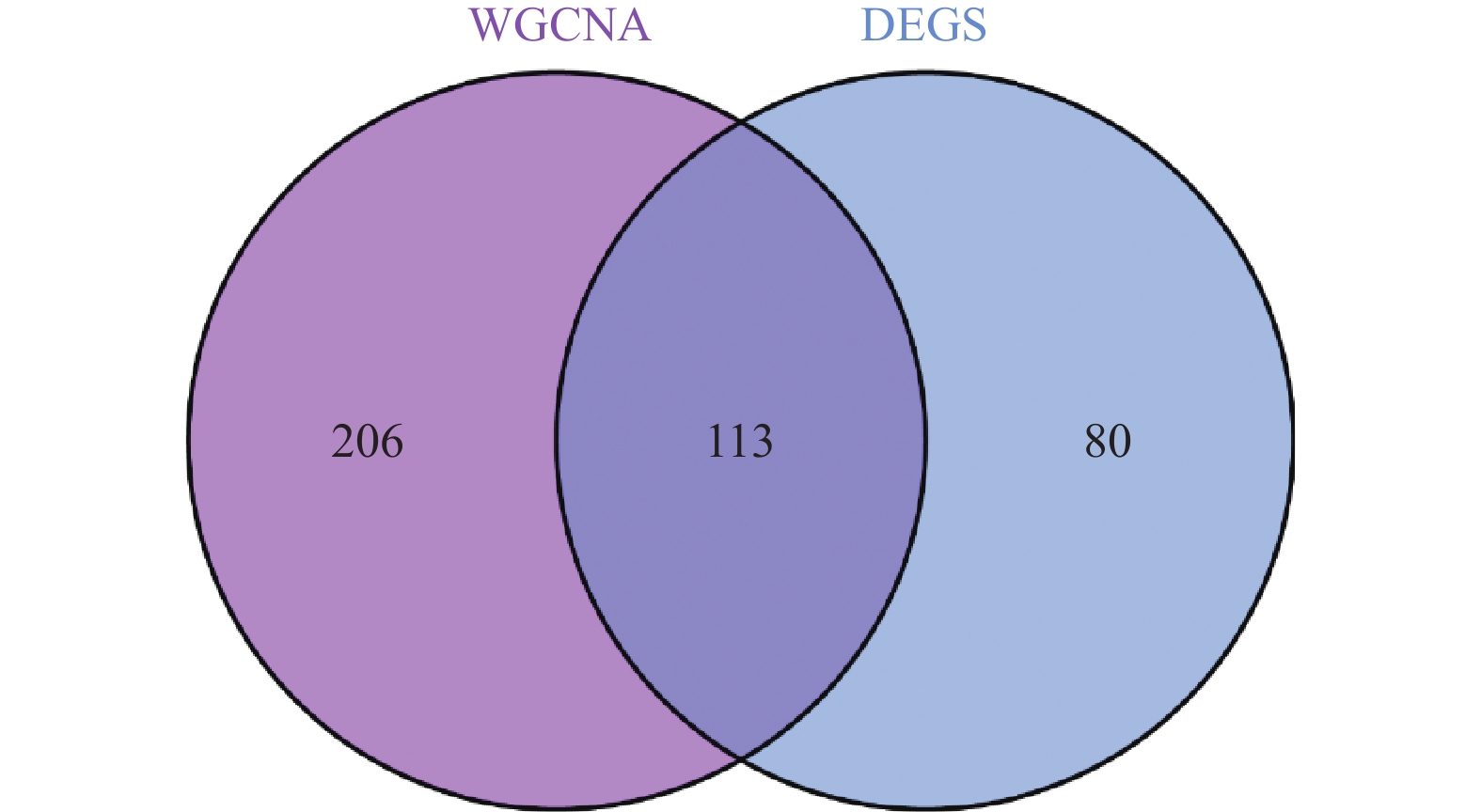
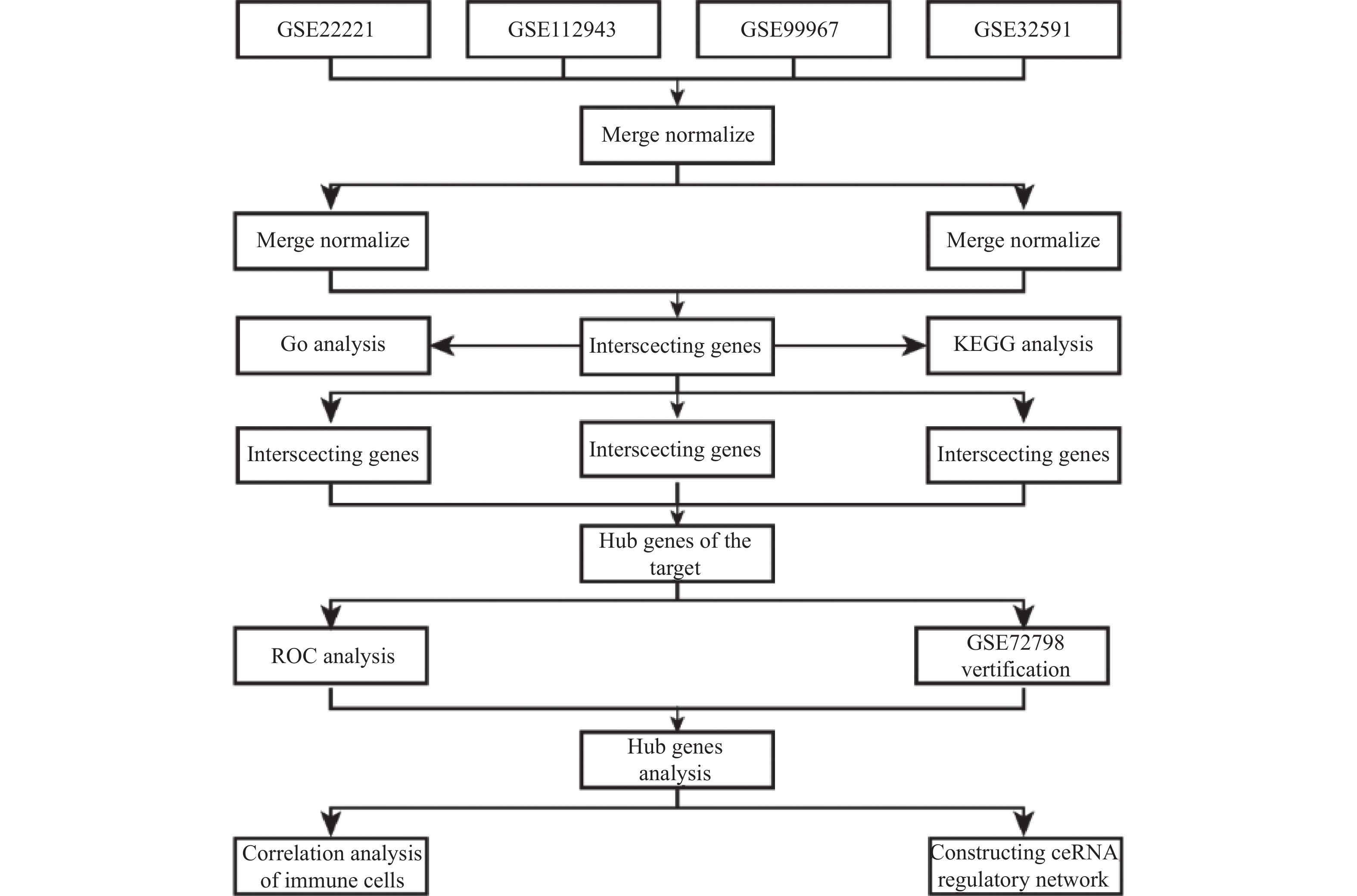
目的 旨在筛选与LN相关的潜在生物标志物,以期用于早期诊断、病情监测和更精准的治疗方案制定。 方法 从基因表达谱数据库(gene expression omnibus,GEO)下载了GSE22221、GSE112943、GSE99967和GSE32591的基因表达数据。通过加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)和微阵列数据的线性模型(linear models for microarray data,LIMMA),获得了交集基因。随后,利用基因本体论(gene ontology,GO)和京都基因与基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)对这些交集基因进行了生物功能和信号通路分析。接着,通过蛋白质-蛋白质相互作用(protein-protein interaction,PPI)网络分析、CytoHubba算法、支持向量机(support vector machine,SVM)和随机森林(random forest,RF)方法,筛选出了与LN高度相关的枢纽基因,进行了受试者工作特征曲线(receiver operating characteristic,ROC)分析,并利用GSE72798数据集对3个潜在的生物标志物进行了验证。 结果 WGCNA分析获得绿黄色模块(P = 7.4e−40)和青色模块(P = 1.5e−14);利用LIMMA法筛选到193个差异表达基因;共鉴定出113个LN相关的交集基因,GO和KEGG分析显示这些基因主要富集在病毒或细菌的防御、I型干扰素信号途径、中性粒细胞介导的免疫和Toll样受体信号等方面。通过CytoHubba、SVM和RF 3种方法筛选出MX1、IFI44和STAT1,其曲线下的面积(area under the curve,AUC)分别为0.874、0.879和0.833。验证数据集显示,MX1、IFI44和STAT1在LN患者中的表达显著高于健康人群(P < 0.001)。 结论 MX1、IFI44和STAT1在LN的发病机制中起到了关键作用,可能成为LN的重要生物标志物和未来的潜在治疗靶点。 Abstract:Objective To identify potential biomarkers associated with LN, with the goal of improving early diagnosis, disease monitoring, and the development of more precise treatment strategies. Methods Gene expression data were downloaded from the Gene Expression Omnibus (GEO) database for datasets GSE22221, GSE112943, GSE99967, and GSE32591. Intersecting genes were obtained through the application of weighted gene co-expression network analysis (WGCNA) and linear models for microarray data (LIMMA). Subsequently, biological function and pathway analyses were conducted on these intersecting genes using Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG). Next, protein-protein interaction (PPI) network analysis was performed, and hub genes highly associated with LN were identified using the CytoHubba algorithm, support vector machine (SVM), and random forest (RF) methods. Receiver operating characteristic (ROC) analysis was performed, and three potential biomarkers were validated using the GSE72798 dataset. Results The green-yellow module (P = 7.4e−40) and the cyan module (P = 1.5e−14) were identified through WGCNA analysis. A total of 193 differentially expressed genes were identified using LIMMA, with 113 intersecting genes related to LN being identified. GO and KEGG analyses indicated that these genes were mainly enriched in viral or bacterial defense, type I interferon signaling pathway, neutrophil-mediated immunity, and Toll-like receptor signaling. MX1, IFI44, and STAT1 were identified as hub genes using CytoHubba, SVM, and RF methods, with AUC values of 0.874, 0.879, and 0.833, respectively. Validation using the GSE72798 dataset demonstrated that the expression of MX1, IFI44, and STAT1 was significantly higher in LN patients compared to healthy individuals (P < 0.001 for all). Conclusion MX1, IFI44, and STAT1 play crucial roles in the pathogenesis of LN and may serve as important biomarkers and potential therapeutic targets for LN. -
Key words:
- Lupus nephritis /
- Systemic lupus erythematosus /
- Biomarker /
- Hub genes /
- Type I interferon
-
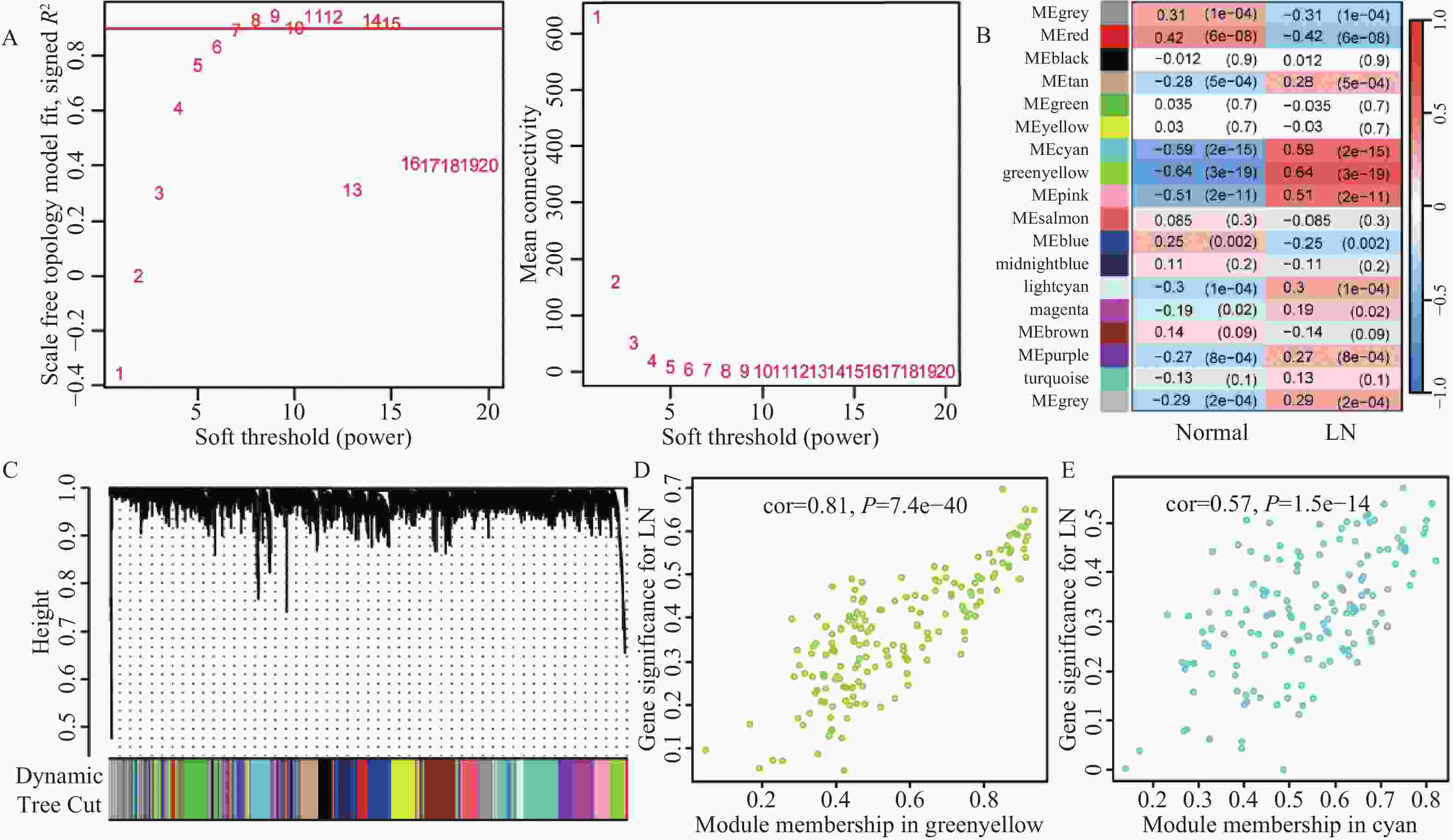
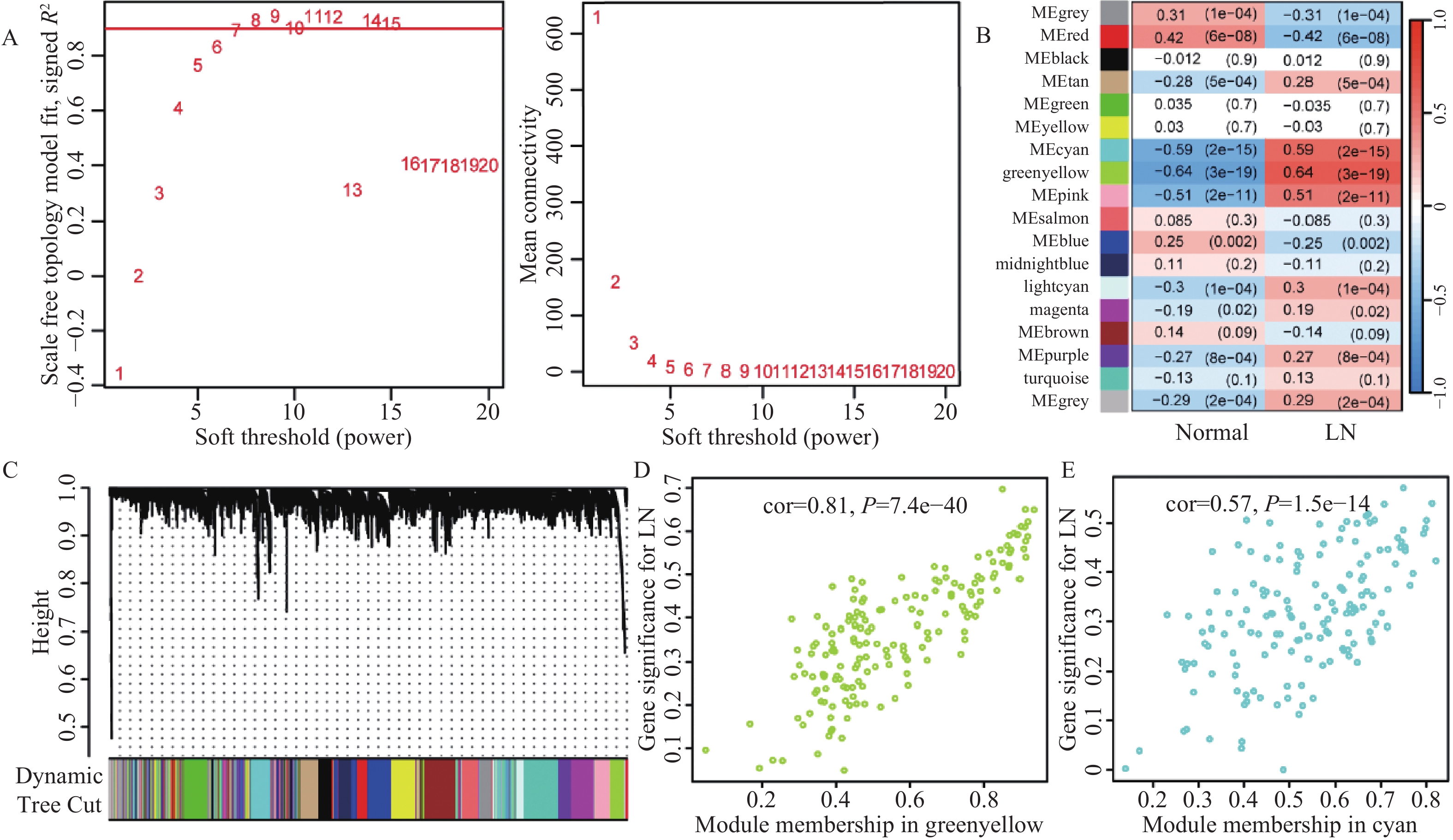
图 3 构建WGCNA网络
A:LN 共表达模块中 power 值筛选;B:模块特征基因与样本特征向量之间的皮尔逊相关系数;C:筛选 LN 的共同表达模块;D:黄绿模块和基因重要性之间的相关性,cor表示 基因显著性(gene significance,GS)和模块成员(module membership,MM)之间的绝对相关系数;E:蓝绿模块和基因重要性之间的相关性,cor表示 GS 和 MM 之间的绝对相关系数。
Figure 3. Construction of the WGCNA network
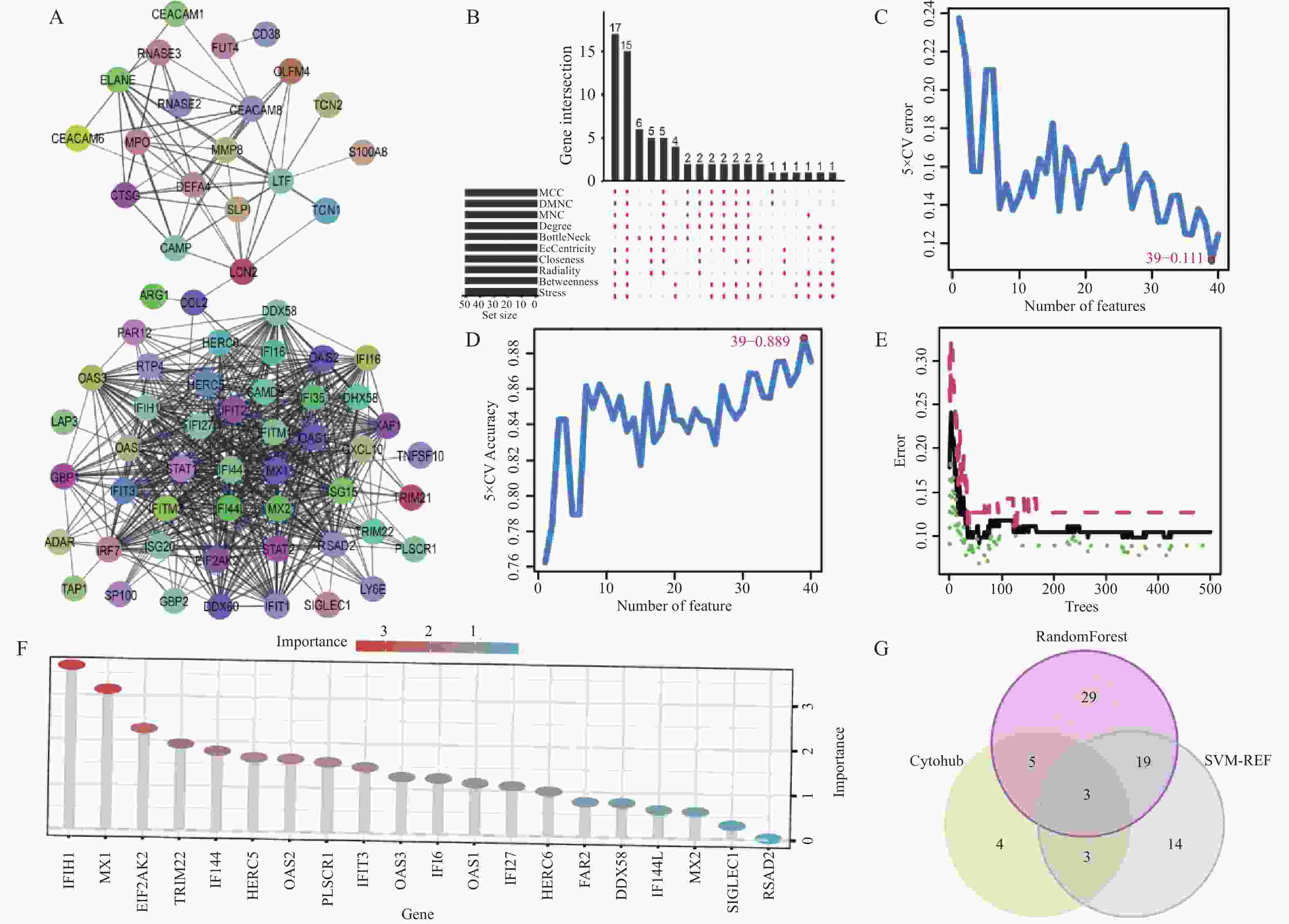
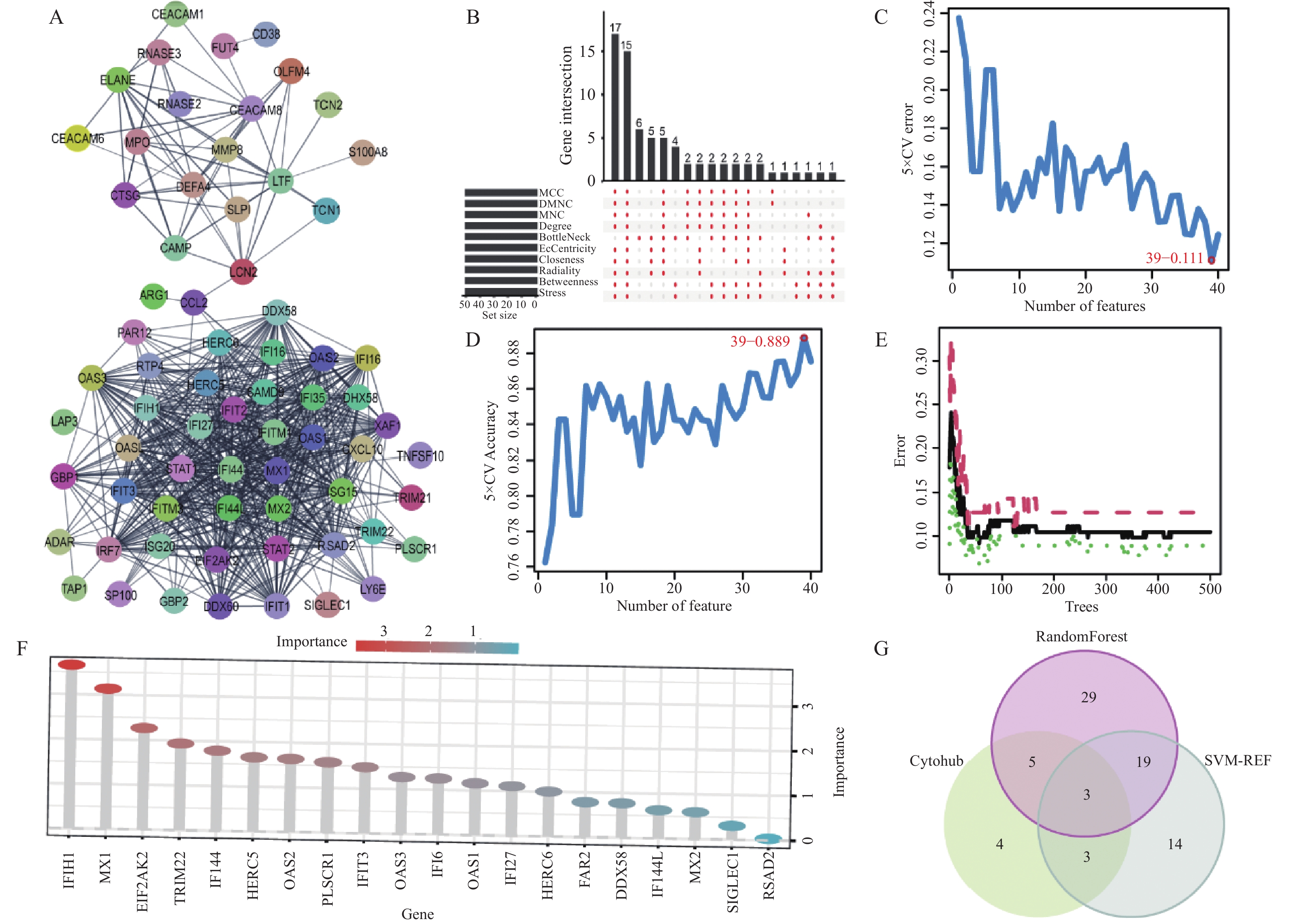
图 5 筛选枢纽基因
A:交集基因的蛋白质互作网络;B:由蛋白质互作网络分析确定的10个拓扑算法的UpSet图;C~D:使用SVM-RFE算法筛选SVM基因;E~F:从交集基因里筛选RandomForest基因及其重要性分析;G:韦恩图显示了3种方法获得的中枢基因的交集。
Figure 5. Screening of hub genes
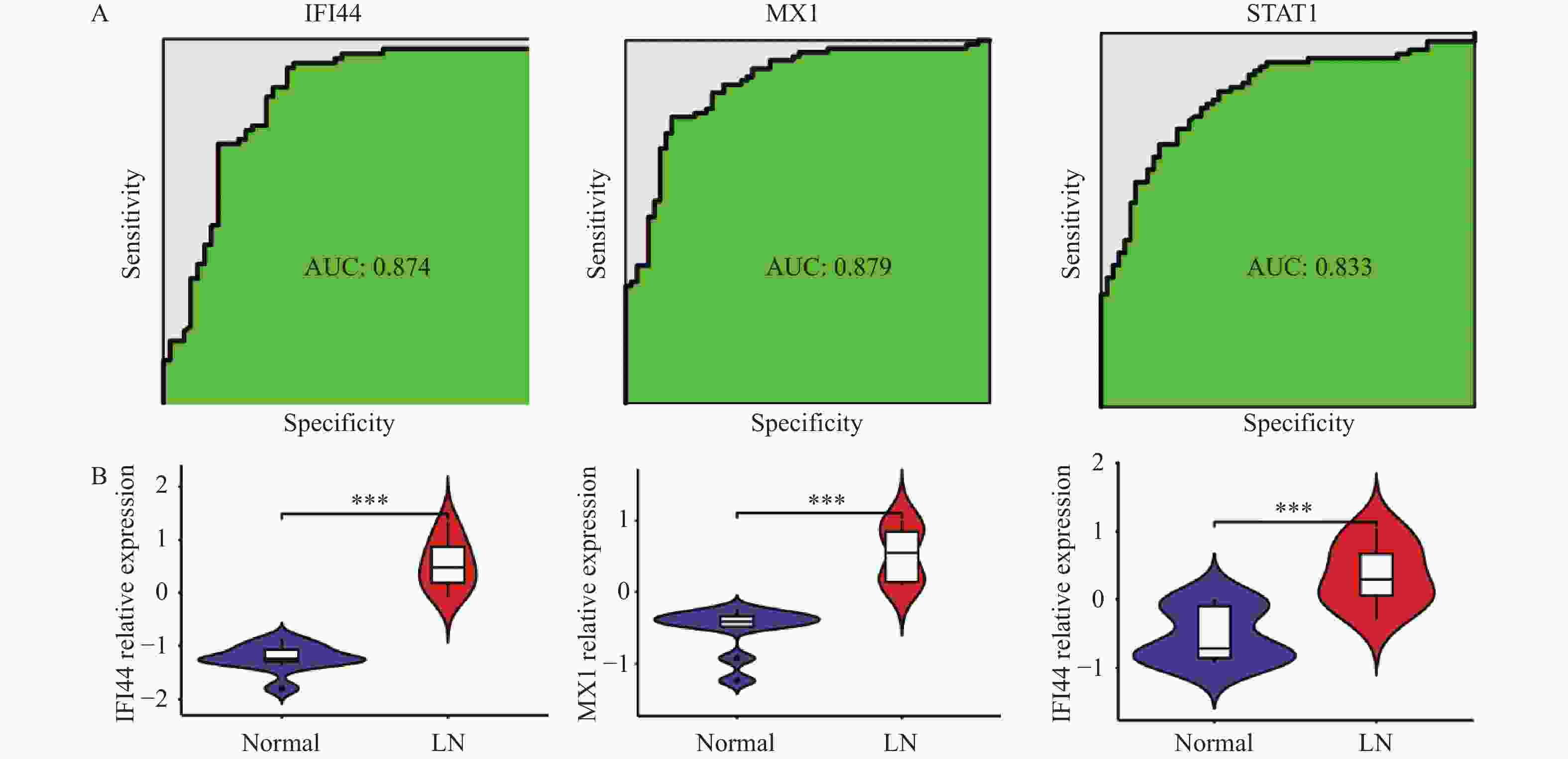
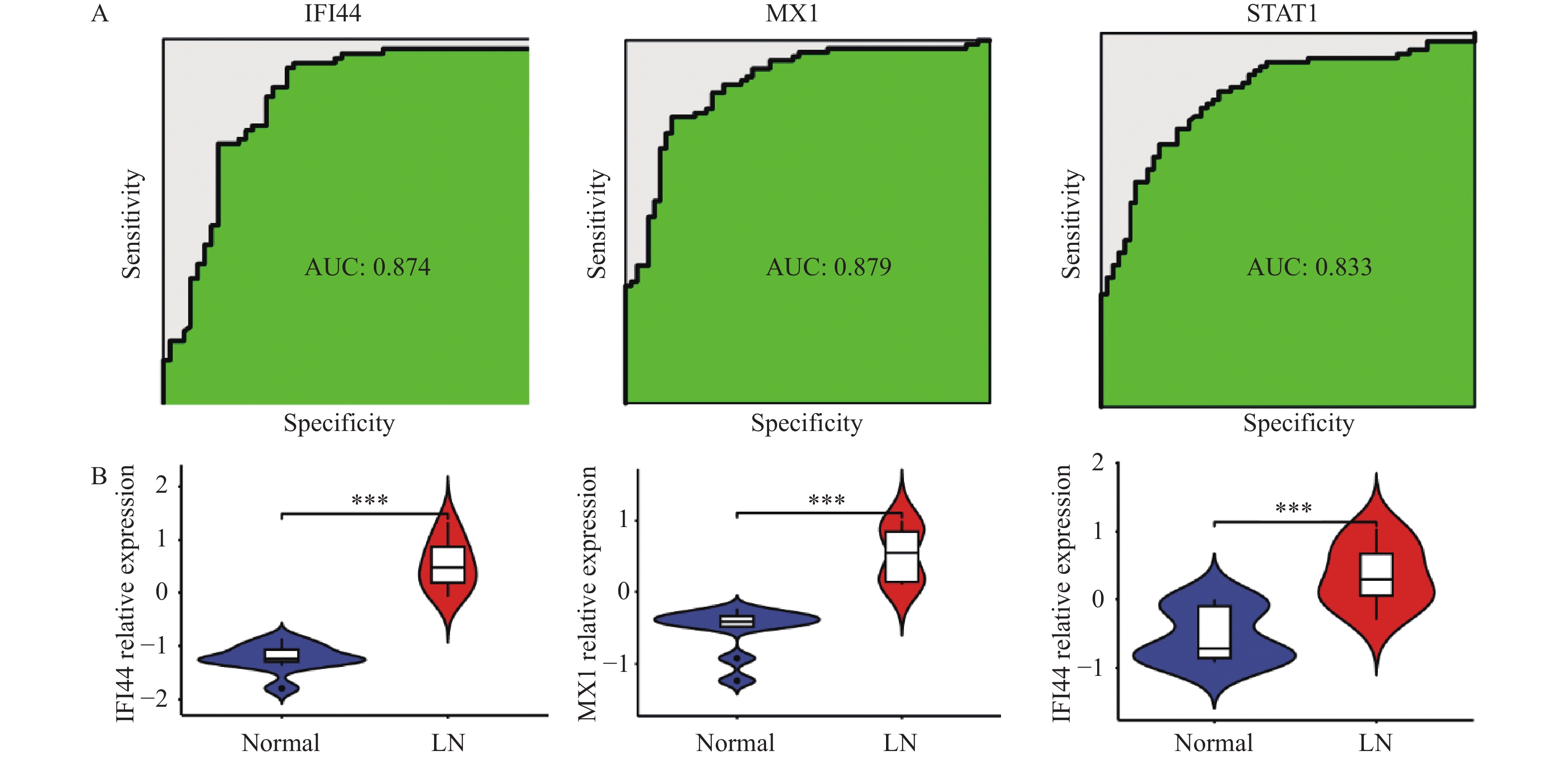
图 6 验证枢纽基因
A:IFI44、MX1和STAT1表达水平的ROC曲线;B:IFI44、MX1和STAT1在验证集中的相对表达量。***P < 0.001。
Figure 6. Validation of hub genes
表 2 正常组与LN患者差异基因
Table 2. Differential genes between normal group and LN patients
差异基因 上调基因 IFI27,IFI44L,IFI44,MX1,LTF,RSAD2,CD163,HERC5,PLSCR1,OAS3,IFIT3,OAS1,OAS2,IFI6,IFIT1,LY96,MX2,HERC6,CREG1,SIGLEC1,EIF2AK2,CEACAM8,C1QB,IFITM1,IFIH1,IFIT2,DDX60,DYSF,ELANE,DEFA4,ABCA1,XAF1,IFITM3,TRIM22,ISG15,LCN2,RNASE6,OLFM4,APOBEC3A,IFI35,RNASE1,VSIG4,MMP9,DHX58,RNASE2,TOP2A,TLR2,LYN,HCK,HBD,MMP8,IRF7,ISG20,ZCCHC2,OLR1,CDC20,GBP2,MS4A4A,DDX58,CCL2,RRM2,SPATS2L,LHFPL2,GBP1,CTSG,CEACAM1,STAT1,CD38,ENTPD1,UBE2J1,TCN2,CXCL10,RTP4,FCER1G,TYMS,B4GALT5,CCNA2,RP2,C3AR1,SAMD9,YIPF6,CARHSP1,TAP1,IFI30,LGALS3BP,TRIM21,FAR2,STIL,PARP12,MFGE8,RNASE3,H2AFZ,CEACAM6,LAP3,YWHAH,GMPR,RIN2,SCARB2,LMNB1,MPO,LCP1,BPGM,TLR4,PRC1,LY6E,CFD,MS4A6A,TPX2,XK,LRRC32,SH3GLB1,TGM2, CTSS,OASL,COL1A2,GINS2,ANP32B,ADAR,RAPGEF5,LYZ,MARCKS,HMGB2,ATP6V1D,SP100,FAM46C,CAMP,COL4A1,TMEM140,ANXA3,FCN1,NETO2,MYOF,EVI2A,FUT4,DNAJC13,CD36,IFI16,POU2AF1,SAMSN1,TCN1,MR1,MNDA,GIMAP4,PRCP,NFE2,CA1,STAT2,PPP1R3D,TACC3,SLPI,C1QA,S100A8,NMI,TLR1,NUSAP1,HLX,ARG1,TMOD1,REXO2,PLA2G4A,PTEN,TNFSF10,NPL,PLEK,GINS3,NOD2,LXN,DNAJC15,HIST1H2AI,MFSD1,DEGS1,FAM46A,CENPN,ITGB2,SCO2,PSMC2,COL15A1,CDC123,DHRS9,HK3,SERPINB1,NOL7,ITGAM 下调基因 CLIC5,ASB1,PTGDS,EPHX2,CDKN1C,TNNC2,KIR2DL3,FCER1A,TGFBR3,FOSB  下载: 导出CSV
下载: 导出CSV
表 3 交集基因GO富集分析
Table 3. GO enrichment analysis of intersection genes
ONTOLOGY ID 描述 P BP GO:0051607 defense response to virus 0.000 GO:0060337 type I interferon signaling pathway 0.000 GO:0042742 defense response to bacterium 0.000 GO:0019221 cytokine-mediated signaling pathway 0.000 GO:0002221 pattern recognition receptor signaling pathway 0.000 GO:0039529 RIG-I signaling pathway 0.000 GO:0002446 neutrophil mediated immunity 0.000 GO:0002709 toll-like receptor signaling pathway 0.000 GO:0002709 regulation of T cell mediated immunity 0.002 CC GO:0042581 specific granule 0.000 GO:0034774 secretory granule lumen 0.000 GO:0060205 cytoplasmic vesicle lumen 0.000 GO:0005766 primary lysosome 0.000 GO:0045335 phagocytic vesicle 0.001 MF GO:0003725 double-stranded RNA binding 0.000 GO:0008301 DNA binding 0.006 GO:0004518 nuclease activity 0.008 GO:0042379 chemokine receptor binding 0.009 GO:0019207 kinase regulator activity 0.015
下载: 导出CSV
表 4 交集基因KEGG信号通路分析
Table 4. KEGG pathway analysis of intersection genes
ID 描述 P hsa05164 Influenza A 0.000 hsa05171 COVID-19 0.000 hsa04621 NOD-like receptor signaling pathway 0.000 hsa04622 RIG-I-like receptor signaling pathway 0.000 hsa05150 Staphylococcus aureus infection 0.001 hsa05322 Systemic lupus erythematosus 0.002 hsa04657 IL-17 signaling pathway 0.004 hsa04623 Cytosolic DNA-sensing pathway 0.016 hsa04610 Complement signaling pathway 0.022 hsa04217 Necroptosis 0.025 hsa04620 Toll-like receptor signaling pathway 0.037 hsa04613 Neutrophil extracellular trap formation 0.044
下载: 导出CSV
-
[1] Bastidas Goyes A R,Mora C,Arsanios D M,et al. Systemic lupus erythematosus,a disease conditioned by the environment[J]. Revista Colombiana de Reumatolog í a,2021,28(6):12-20. [2] Barber M R W,Drenkard C,Falasinnu T,et al. Global epidemiology of systemic lupus erythematosus[J]. Nat Rev Rheumatol,2021,17(9):515-532. [3] Gasparotto M,Gatto M,Binda V,et al. Lupus nephritis: Clinical presentations and outcomes in the 21st centu ry[J]. Rheumatology (Oxford),2020,59(Suppl5):v39-v51. [4] Heo YA. Voclosporin: First approval[J]. Drugs,2021,81(5):605-610. [5] Mejia-Vilet J M,Malvar A,Arazi A,et al. The lupus nephritis management renaissance[J]. Kidney Int,2022,101(2):242-255. doi: 10.1016/j.kint.2021.09.012 [6] Parikh S V,Almaani S,Brodsky S,et al. Update on lupus nephritis: Core curriculum 2020[J]. American Journal of Kidney Diseases,2020,76(2):265-281. doi: 10.1053/j.ajkd.2019.10.017 [7] Bernard L,Wang A R,Menez S,et al. Kidney biopsy utility: Patient and clinician perspectives from the kidney precision medicine project[J]. Kidney Med,2023,5(10):100707-100715. doi: 10.1016/j.xkme.2023.100707 [8] Satam H,Joshi K,Mangrolia U,et al. Next-generation sequencing technology: Current trends and advancements[J]. Biology (Basel),2023,12(7):997-1021. doi: 10.3390/biology12070997 [9] Langfelder P,Horvath S. Wgcna: An r package for weighted correlation network analysis[J]. BMC Bioinformatics,2008,9(12):559-571. [10] Nedaie A,Najafi A A. Support vector machine with dirichlet feature mapping[J]. Neural Netw,2017,98(2):87-101. [11] Ubels J,Schaefers T,Punt C,et al. Rainforest: A random forest approach to predict treatment benefit in data from (failed) clinical drug trials[J]. Bioinformatics,2020,36(Suppl_2):i601-i609. [12] Morris A C,Forbes-Osborne M A,Pillai L S,et al. Microarray analysis of xops-mcfp zebrafish retina identifies genes associated with rod photoreceptor degeneration and regeneration[J]. Invest Ophthalmol Vis Sci,2011,52(5):2255-2266. doi: 10.1167/iovs.10-6022 [13] Ko WC C,Li L,Young T R,et al. Gene expression profiling in the skin reveals strong similarities between subacute and chronic cutaneous lupus that are distinct from lupus nephritis[J]. J Invest Dermatol,2021,141(12):2808-2819. doi: 10.1016/j.jid.2021.04.030 [14] Wither J E,Prokopec S D,Noamani B,et al. Identification of a neutrophil-related gene expression signature that is enriched in adult systemic lupus erythematosus patients with active nephritis: Clinical/pathologic associations and etiologic mechanisms[J]. PLoS One,2018,13(5):e0196117. doi: 10.1371/journal.pone.0196117 [15] Berthier C C,Bethunaickan R,Gonzalez-Rivera T,et al. Cross-species transcriptional network analysis defines shared inflammatory responses in murine and human lupus nephritis[J]. J Immunol,2012,189(2):988-1001. [16] Ducreux J,Houssiau F A,Vandepapelière P,et al. Interferon α kinoid induces neutralizing anti-interferon α antibodies that decrease the expression of interferon-induced and b cell activation associated transcripts: Analysis of extended follow-up data from the interferon α kinoid phase i/ii study[J]. Rheumatology (Oxford),2016,55(10):1901-1905. doi: 10.1093/rheumatology/kew262 [17] McKellar J,Moncorgé O,Goujon C. Mx1 proteins and influenza a virus under the microscope[J]. Virologie (Montrouge),2023,27(1):31-34. doi: 10.1684/vir.2023.0990 [18] Tao S,Tan X,Chai W,et al. Knockdown of klf5 ameliorates renal fibrosis in mrl/lpr mice via inhibition of mx1 transcription[J]. Immun Inflamm Dis,2023,11(7):e937. doi: 10.1002/iid3.937 [19] Feng X,Wu H,Grossman J M,et al. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus eryt hematosus[J]. Arthritis Rheum,2006,54(9):2951-2962. [20] Busse D C,Habgood-Coote D,Clare S,et al. Interferon-induced protein 44 and interferon-induced protein 44-like restrict replication of respiratory syncytial virus[J]. J Virol,2020,94(18):e00297. [21] DeDiego M L,Nogales A,Martinez-Sobrido L,et al. Interferon-induced protein 44 interacts with cellular fk506-binding protein 5,negatively regulates host antiviral responses,and supports virus replication[J]. mBio,2019,10(4):e01839. [22] Philips R L,Wang Y,Cheon H,et al. The jak-stat pathway at 30: Much learned,much more to do[J]. Cell,2022,185(21):3857-3876. doi: 10.1016/j.cell.2022.09.023 [23] Xu W,Chen Z,Shen X,et al. Reno-protective effect of realgar nanoparticles on lupus nephritis of mrl/lpr mice through stat1[J]. Iran J Immunol,2019,16(2):170-181. [24] Fu Y,Xiang Y,Wang Y,et al. The stat1/hmgb1/nf-κb pathway in chronic inflammation and kidney injury after cisplatin exposure[J]. Theranostics,2023,13(9):2757-2773. doi: 10.7150/thno.81406 [25] Kemmner S,Bachmann Q,Steiger S,et al. Stat1 regulates macrophage number and phenotype and prevents renal fibrosis after ischemia-reperfusion injury[J]. American Journal of Physiology-Renal Physiology,2019,316(2):F277-F291. [26] Ding X,Ren Y,He X. Ifn-i mediates lupus nephritis from the beginning to renal fibrosis[J]. Front Immunol,2021,12(4):676082-676099. [27] Arazi A,Rao D A,Berthier C C,et al. The immune cell landscape in kidneys of patients with lupus nephritis[J]. Nature immunology,2019,20(7):902-914. -
 点击查看大图
点击查看大图
计量
- 文章访问数: 2006
- HTML全文浏览量: 1346
- PDF下载量: 19
- 被引次数: 0
